Phylogeny of Salmonids (Salmoniformes: Salmonidae) and Its Molecular Dating: Analysis of Mtdna Data S
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Status of the Endangered Freshwater Fishes in China and the Analysis of the Endangered Causes Institute of Hydrobiology
The status of the endangered freshwater fishes in China and The analysis of the endangered causes HE Shunping, CIIEN Yiyu Institute of Hydrobiology, CAS, Wuhan, ITubei Province, 430072 Abstract More than 800 species of freshwater fishes are precious biological resources in inland water system of China. Among them, there are a great number of endemic and precious group, and a lot of monotypic genera and species. Recently, owing to the synthetic effects of the natural and human-beings, many of these fishes gradually became endangered. The preliminary statistic result indicates that 92 species are endangered fishes and account for 10% of the total freshwater fishes in China. For the purpose of protection of the biodiversity of fishes, it is necessary to analyse these causes which have led the fishes to become endangered. This report could be used as a scientific reference for researching and saving the endemic precious freshwater fishes in China. Key words Endangered freshwater fishes, Endangered causes, China In the process of the evolution of living things, along with the origin of life, the extinction of life also existed. In the long_ life history, the speciation and the extinction of living things often keep a relative balance. As time goes on, especially after by the impact of human beings activity of production and life, the pattern of the biodiversity were changed or damaged, more or less. At last, in the modern society, human beings activity not only accelerate the progress of society and the development of economy, but also, as a special species, become the source of disturbing_ to other species. -

Fresh- and Brackish-Water Cold-Tolerant Species of Southern Europe: Migrants from the Paratethys That Colonized the Arctic
water Review Fresh- and Brackish-Water Cold-Tolerant Species of Southern Europe: Migrants from the Paratethys That Colonized the Arctic Valentina S. Artamonova 1, Ivan N. Bolotov 2,3,4, Maxim V. Vinarski 4 and Alexander A. Makhrov 1,4,* 1 A. N. Severtzov Institute of Ecology and Evolution, Russian Academy of Sciences, 119071 Moscow, Russia; [email protected] 2 Laboratory of Molecular Ecology and Phylogenetics, Northern Arctic Federal University, 163002 Arkhangelsk, Russia; [email protected] 3 Federal Center for Integrated Arctic Research, Russian Academy of Sciences, 163000 Arkhangelsk, Russia 4 Laboratory of Macroecology & Biogeography of Invertebrates, Saint Petersburg State University, 199034 Saint Petersburg, Russia; [email protected] * Correspondence: [email protected] Abstract: Analysis of zoogeographic, paleogeographic, and molecular data has shown that the ancestors of many fresh- and brackish-water cold-tolerant hydrobionts of the Mediterranean region and the Danube River basin likely originated in East Asia or Central Asia. The fish genera Gasterosteus, Hucho, Oxynoemacheilus, Salmo, and Schizothorax are examples of these groups among vertebrates, and the genera Magnibursatus (Trematoda), Margaritifera, Potomida, Microcondylaea, Leguminaia, Unio (Mollusca), and Phagocata (Planaria), among invertebrates. There is reason to believe that their ancestors spread to Europe through the Paratethys (or the proto-Paratethys basin that preceded it), where intense speciation took place and new genera of aquatic organisms arose. Some of the forms that originated in the Paratethys colonized the Mediterranean, and overwhelming data indicate that Citation: Artamonova, V.S.; Bolotov, representatives of the genera Salmo, Caspiomyzon, and Ecrobia migrated during the Miocene from I.N.; Vinarski, M.V.; Makhrov, A.A. -

The Arctic Char (Salvelinus Alpinus) “Complex” in North America Revisited
The Arctic char (Salvelinus alpinus) “complex” in North America revisited Eric B. Taylor Hydrobiologia The International Journal of Aquatic Sciences ISSN 0018-8158 Hydrobiologia DOI 10.1007/s10750-015-2613-6 1 23 Author's personal copy Hydrobiologia DOI 10.1007/s10750-015-2613-6 CHARR II Review Paper The Arctic char (Salvelinus alpinus) ‘‘complex’’ in North America revisited Eric B. Taylor Received: 1 July 2015 / Revised: 16 November 2015 / Accepted: 5 December 2015 Ó Springer International Publishing Switzerland 2015 Abstract The Arctic char (Salvelinus alpinus) law. This research has significantly revised what species ‘‘complex’’ has fascinated biologists for constitutes the S. alpinus species ‘‘complex’’, provided decades particularly with respect to how many species insights into the ecology and genetics of co-existence, there are and their geographic distributions. I review and promoted conservation assessment that better recent research on the species complex, focussing on represents biodiversity within Salvelinus. A geograph- biodiversity within northwestern North America, ically and genetically comprehensive analysis of which indicates (i) what was once considered a single relationships among putative taxa of Pan-Pacific taxon consists of three taxa: S. alpinus (Arctic char), S. Salvelinus is still required to better quantify the malma (Dolly Varden), and S. confluentus (bull trout), number of taxa and their origins. (ii) morphological and genetic data indicate that S. alpinus and S. malma, and S. malma and S. confluentus Keywords Dolly Varden Á Arctic char Á Bull trout Á exist as distinct biological species in sympatry, (iii) Geographic distribution Á Taxonomy Á Conservation sympatric forms of S. alpinus exist in Alaska as in other areas of the Holarctic, (iv) Dolly Varden comprises two well-differentiated subspecies, S. -

Chromosomal Study of the Lenoks, Brachymystax(Salmoniformes
Journal of Species Research 2(1):91-98, 2013 Chromosomal study of the lenoks, Brachymystax (Salmoniformes, Salmonidae) from the South of the Russian Far East I.V. Kartavtseva*, L.K. Ginatulina, G.A. Nemkova and S.V. Shedko Institute of Biology and Soil Science of the Far East Branch of the Russian Academy of Sciences, Prospect 100 let Vladivostoku 159, Vladivostok 690022 *Correspondent: [email protected], [email protected] An investigation of the karyotypes of two species of the genus Brachymystax (B. lenok and B. tumensis) has been done for the Russia Primorye rivers running to the East Sea basin, and others belonging to Amur basin. Based on the analysis of two species chromosome characteristics, combined with original and literary data, four cytotypes have been described. One of these cytotypes (Cytotype I: 2n=90, NF=110-118) was the most common. This common cytotype belongs to B. tumensis from the rivers of the East Sea basin and B. lenok from the rivers of the Amur basin, i.e. extends to the zones of allopatry. In the rivers of the Amur river basin, in the zone of the sympatric habitat of two species, each taxon has karyotypes with different chromosome numbers, B. tumensis (2n=92) and B. lenok (2n=90). Because of the ability to determine a number of the chromosome arms for these two species, additional cytotype have been identified for B. tum- ensis: Cytotype II with 2n=92, NF=110-124 in the rivers basins of the Yellow sea and Amur river and for B. lenok three cytotypes: Cytotype I: 2n=90, NF=110 in the Amur river basin; Cytotype III with 2n=90, NF=106-126 in the Amur river basin and Cytotypes IV with 2n=92, NF=102 in the Baikal lake. -
Qrno. 1 2 3 4 5 6 7 1 CP 2903 77 100 0 Cfcl3
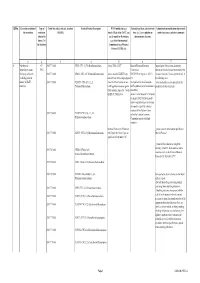
QRNo. General description of Type of Tariff line code(s) affected, based on Detailed Product Description WTO Justification (e.g. National legal basis and entry into Administration, modification of previously the restriction restriction HS(2012) Article XX(g) of the GATT, etc.) force (i.e. Law, regulation or notified measures, and other comments (Symbol in and Grounds for Restriction, administrative decision) Annex 2 of e.g., Other International the Decision) Commitments (e.g. Montreal Protocol, CITES, etc) 12 3 4 5 6 7 1 Prohibition to CP 2903 77 100 0 CFCl3 (CFC-11) Trichlorofluoromethane Article XX(h) GATT Board of Eurasian Economic Import/export of these ozone destroying import/export ozone CP-X Commission substances from/to the customs territory of the destroying substances 2903 77 200 0 CF2Cl2 (CFC-12) Dichlorodifluoromethane Article 46 of the EAEU Treaty DECISION on August 16, 2012 N Eurasian Economic Union is permitted only in (excluding goods in dated 29 may 2014 and paragraphs 134 the following cases: transit) (all EAEU 2903 77 300 0 C2F3Cl3 (CFC-113) 1,1,2- 4 and 37 of the Protocol on non- On legal acts in the field of non- _to be used solely as a raw material for the countries) Trichlorotrifluoroethane tariff regulation measures against tariff regulation (as last amended at 2 production of other chemicals; third countries Annex No. 7 to the June 2016) EAEU of 29 May 2014 Annex 1 to the Decision N 134 dated 16 August 2012 Unit list of goods subject to prohibitions or restrictions on import or export by countries- members of the -

Evolutionary Genomics of a Plastic Life History Trait: Galaxias Maculatus Amphidromous and Resident Populations
EVOLUTIONARY GENOMICS OF A PLASTIC LIFE HISTORY TRAIT: GALAXIAS MACULATUS AMPHIDROMOUS AND RESIDENT POPULATIONS by María Lisette Delgado Aquije Submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy at Dalhousie University Halifax, Nova Scotia August 2021 Dalhousie University is located in Mi'kma'ki, the ancestral and unceded territory of the Mi'kmaq. We are all Treaty people. © Copyright by María Lisette Delgado Aquije, 2021 I dedicate this work to my parents, María and José, my brothers JR and Eduardo for their unconditional love and support and for always encouraging me to pursue my dreams, and to my grandparents Victoria, Estela, Jesús, and Pepe whose example of perseverance and hard work allowed me to reach this point. ii TABLE OF CONTENTS LIST OF TABLES ............................................................................................................ vii LIST OF FIGURES ........................................................................................................... ix ABSTRACT ...................................................................................................................... xii LIST OF ABBREVIATION USED ................................................................................ xiii ACKNOWLEDGMENTS ................................................................................................ xv CHAPTER 1. INTRODUCTION ....................................................................................... 1 1.1 Galaxias maculatus .................................................................................................. -

Revalidation and Redescription of Brachymystax Tsinlingensis Li, 1966 (Salmoniformes: Salmonidae) from China
Zootaxa 3962 (1): 191–205 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2015 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3962.1.12 http://zoobank.org/urn:lsid:zoobank.org:pub:F7864FFE-F182-455E-B37A-8A253D8DB72D Revalidation and redescription of Brachymystax tsinlingensis Li, 1966 (Salmoniformes: Salmonidae) from China YING-CHUN XING1,2, BIN-BIN LV3, EN-QI YE2, EN-YUAN FAN1, SHI-YANG LI4, LI-XIN WANG4, CHUN- GUANG ZHANG2,* & YA-HUI ZHAO2,* 1Natural Resource and Environment Research Center, Chinese Academy of Fishery Sciences, Beijing, China. 2Institute of Zoology, Chinese Academy of Sciences, Beijing, China. 3Yellow River Fisheries Research Institute, Chinese Academy of Fishery Sciences, Xi’an, China. 4The College of Forestry of Beijing Forestry University, Beijing, China. *Corresponding authors: Yahui Zhao, [email protected]; Chunguang Zhang, [email protected] Abstract Brachymystax tsinlingensis Li, 1966 is revalidated and redescribed. It can be distinguished from all congeners by the fol- lowing combination of characteristics: no spots on operculum; gill rakers 15-20; lateral-line scales 98-116; pyloric caeca 60-71. Unique morphological characters and genetic divergence of this species are discussed. This species has a limited distribution in several streams of the middle part of the Qinling Mountains in China. Methods for management and pro- tection of B. tsinlingensis need to be re-evaluated. Key words: Brachymystax, revalidation, redescription, Salmonidae, China Introduction The genus Brachymystax Günther, 1866, belonging to Salmonidae, Salmoniformes, is distributed in eastern and northern Asia with three currently recognized valid species (Froese & Pauly, 2014): B. -

Holarctic Phylogeography of Arctic Charr (Salvelinus Alpinus L.) Inferred from Mitochondrial Dna Sequences
Evolution, 55(3), 2001, pp. 573±586 HOLARCTIC PHYLOGEOGRAPHY OF ARCTIC CHARR (SALVELINUS ALPINUS L.) INFERRED FROM MITOCHONDRIAL DNA SEQUENCES PATRICK C. BRUNNER,1,2,5 MARLIS R. DOUGLAS,1 ALEXANDER OSINOV,3 CHRIS C. WILSON,4 AND LOUIS BERNATCHEZ2 1Zoologisches Museum, UniversitaÈt ZuÈrich, Winterthurerstrasse 190, CH-8057 ZuÈrich, Switzerland 2DeÂpartement de biologie, GIROQ, Universite Laval, Saint-Foy QueÂbec G1K 7P4, Canada 3Department of Ichthyology, Moscow State University, Moscow, 119899, Russia 4Ontario Ministry of Natural Resources, Aquatic Ecosystems Science Section, Trent University, Peterborough Ontario, K9J 8N8, Canada Abstract. This study evaluated mitochondrial DNA (mtDNA) sequence variation in a 552-bp fragment of the control region of Arctic charr (Salvelinus alpinus) by analyzing 159 individuals from 83 populations throughout the entire range of the complex. A total of 89 (16.1%) nucleotide positions were polymorphic, and these de®ned 63 haplotypes. Phylogenetic analyses supported the monophyly of the complex and assigned the observed haplotypes to ®ve geographic regions that may be associated with different glacial refugia. Most notably, a formerly de®ned major evolutionary lineage (S. a. erythrinus) ranging from North America across the Arctic archipelago to the Eurasian continent has now been partitioned into the Arctic group and the newly identi®ed Siberian group. The Beringian group, formed entirely by specimens assigned to S. malma (Dolly Varden), encompassed the area formerly assigned to S. a. taranetzi. The latter, due to a unique haplotype, became the basal member of the Arctic group. Overall, the S. alpinus complex re¯ects divergent evolutionary groups coupled with shallow intergroup differentiation, also indicated by an analysis of molecular variance that attributed 73.7% (P , 0.001) of the total genetic variance among groups. -

Distribution and Heterogeneity of Heterochromatin in the European Huchen
PL-ISSN0015-5497(print),ISSN1734-9168(online) FoliaBiologica(Kraków),vol.62(2014),No2 Ó InstituteofSystematicsandEvolutionofAnimals,PAS,Kraków, 2014 doi:10.3409/fb62_2.81 DistributionandHeterogeneityofHeterochromatinintheEuropean Huchen(Huchohucho Linnaeus,1758)(Salmonidae)* KUCINSKI Marcin,OCALEWICZ Konrad,FOPP-BAYAT Dorota,LISZEWSKI Tomasz, FURGALA-SELEZNIOW Grazyna,JANKUN Malgorzata AcceptedFebruary19,2014 KUCINSKI M., OCALEWICZ K., FOPP-BAYAT D., LISZEWSKI T., FURGALA-SELEZNIOW G., JANKUN M. 2014. Distribution and heterogeneity of heterochromatinintheEuropeanhuchen (Hucho hucho Linnaeus, 1758) (Salmonidae). Folia Biologica (Kraków) 62: 81-89. The chromosomal characteristics, locations and variations of the heterochromatin were studied in the European huchen (Hucho hucho, Linnaeus, 1758) karyotype using conventional C- banding, endonuclease digestion banding, silver nitrate (AgNO3), chromomycin A3 (CMA3) and DAPIstainingtechniques.Thekaryotypeconsistsof82 chromosomes: 13 pairs of metacentric chromosomes, 2 pairs of submetacentric chromosomes and 26 pairs of subtelo-acrocentric chromosomes (NF=112). Original data on the chromosomal distribution of segments resistant to Alu I, Dde I and Mbo I restriction endonucleases and identification of the C-banded heterochromatin presented here have been used to characterize the huchen karyotype. On the basis of the banding patterns provided in the course of restriction enzyme digestion, AgNO3/CMA3 staining and C-banding we distinguished twelve types of heterochromatin grouped in four areas of the -

Preparing the Shaanxi-Qinling Mountains Integrated Ecosystem Management Project (Cofinanced by the Global Environment Facility)
Technical Assistance Consultant’s Report Project Number: 39321 June 2008 PRC: Preparing the Shaanxi-Qinling Mountains Integrated Ecosystem Management Project (Cofinanced by the Global Environment Facility) Prepared by: ANZDEC Limited Australia For Shaanxi Province Development and Reform Commission This consultant’s report does not necessarily reflect the views of ADB or the Government concerned, and ADB and the Government cannot be held liable for its contents. (For project preparatory technical assistance: All the views expressed herein may not be incorporated into the proposed project’s design. FINAL REPORT SHAANXI QINLING BIODIVERSITY CONSERVATION AND DEMONSTRATION PROJECT PREPARED FOR Shaanxi Provincial Government And the Asian Development Bank ANZDEC LIMITED September 2007 CURRENCY EQUIVALENTS (as at 1 June 2007) Currency Unit – Chinese Yuan {CNY}1.00 = US $0.1308 $1.00 = CNY 7.64 ABBREVIATIONS ADB – Asian Development Bank BAP – Biodiversity Action Plan (of the PRC Government) CAS – Chinese Academy of Sciences CASS – Chinese Academy of Social Sciences CBD – Convention on Biological Diversity CBRC – China Bank Regulatory Commission CDA - Conservation Demonstration Area CNY – Chinese Yuan CO – company CPF – country programming framework CTF – Conservation Trust Fund EA – Executing Agency EFCAs – Ecosystem Function Conservation Areas EIRR – economic internal rate of return EPB – Environmental Protection Bureau EU – European Union FIRR – financial internal rate of return FDI – Foreign Direct Investment FYP – Five-Year Plan FS – Feasibility -

Current Global Status of Taimen and the Need to Implement Aggressive Conservation Measures to Avoid Population and Species-Level Extinction
Arch. Pol. Fish. (2013) 21: 119-128 DOI 10.2478/aopf-2013-0009 RESEARCH ARTICLE Current global status of taimen and the need to implement aggressive conservation measures to avoid population and species-level extinction Peter S. Rand Received – 06 April 2013/Accepted – 10 September 2013. Published online: 30 September 2013; ©Inland Fisheries Institute in Olsztyn, Poland Citation: Rand P.S. 2013 – Current global status of taimen and the need to implement aggressive conservation measures to avoid population and species-level extinction – Arch. Pol. Fish. 21: 119-128. Abstract. An international effort carried out during Introduction 2011-2012 culminated in three completed IUCN status assessments of Hucho spp., thus completing assessments for all species in this and a related genus Parahucho. These Fishes are grossly underrepresented on the IUCN species hold great ecological and evolutionary significance in Red List of Threatened Species. Recent efforts are in- the salmonid family, and are recognized as the largest tended to rectify this problem (Kottelat and Freyhof salmonids in the world. Specialists have long recognized their 2007, Tweddle et al. 2009). There is a clear need to precarious status. The reports conclude that all species of taimen are now listed as threatened or Data Deficient on The better assess freshwater taxa in general considering IUCN Red List of Threatened Species™, and point to a host of the disproportionate effect that humans have on ongoing and emerging threats, including habitat loss and freshwater habitat (Vörösmarty et al. 2010). overharvest. I summarize the key data used to arrive at the status categories, and emphasize some key conservation The species in the genera Hucho and Parahucho measures that must be taken to avoid extinction at both the serve as the classic conservation flagship species, local population scale and at the species level. -

Arctic Biodiversity Synthesis
ACSAO-SE04 Stockholm Doc 3.5b Mar 2013 ABA Synthesis Final draft ABA Synthesis – February 20th 2013 Chapter 1 Arctic Biodiversity Synthesis Authors Hans Meltofte, Tom Barry, Dominique Berteaux, Helga Bü ltmann, Jørgen S. Christiansen, Joseph A. Cook, Anders Dahlberg, Fred J.A. Daniëls, Dorothee Ehrich, Finnur Friðriksson, Barbara Ganter, Anthony J. Gaston, Lynn Gillespie, Lenore Grenoble, Eric P. Hoberg, Ian D. Hodkinson, Henry P. Huntington, Rolf A. Ims, Alf B. Josefson, Susan J. Kutz, Sergius L. Kuzmin, Kristin L. Laidre, Dennis R. Lassuy, Patrick N. Lewis, Connie Lovejoy, Christine Michel, Vadim Mokievsky, Tero Mustonen, David C. Payer, Michel Poulin, Donald Reid, James D. Reist, David F. Tessler and Frederick J. Wrona ”Nowadays all of the tundra is on the move now. Many forest animals are coming to tundra now. Moose is moving towards the tundra proper nowadays” (Alexey Nikolayevich Kemlil, a Chukchi reindeer herder from Turvaurgin in northeastern Sakha-Yakutia, Siberia; T. Mustonen in litt.). “I too, have noticed changes to the climate in our area. It has progressed with frightening speed especially the last few years. In Iqaluktutiaq, the landscape has changed. The land is now a stranger, it seems, based on our accumulated knowledge. The seasons have shifted, the ice is thinner and weaker, and the streams, creeks and rivers have changed their characteristics” (Analok, Cambridge Bay, Victoria Island, Nunavut; Elders Conference on Climate Change 2001). Photo caption Climate change is already causing earlier snow melt, which initially may benefit many Arctic organisms. But in the longer term it will make it possible for more competitive southern species to ‘take over’ what are currently Arctic habitats.