(12) Patent Application Publication (43) Pub. Date: Aug
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Classification of Medicinal Drugs and Driving: Co-Ordination and Synthesis Report
Project No. TREN-05-FP6TR-S07.61320-518404-DRUID DRUID Driving under the Influence of Drugs, Alcohol and Medicines Integrated Project 1.6. Sustainable Development, Global Change and Ecosystem 1.6.2: Sustainable Surface Transport 6th Framework Programme Deliverable 4.4.1 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Due date of deliverable: 21.07.2011 Actual submission date: 21.07.2011 Revision date: 21.07.2011 Start date of project: 15.10.2006 Duration: 48 months Organisation name of lead contractor for this deliverable: UVA Revision 0.0 Project co-funded by the European Commission within the Sixth Framework Programme (2002-2006) Dissemination Level PU Public PP Restricted to other programme participants (including the Commission x Services) RE Restricted to a group specified by the consortium (including the Commission Services) CO Confidential, only for members of the consortium (including the Commission Services) DRUID 6th Framework Programme Deliverable D.4.4.1 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Page 1 of 243 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Authors Trinidad Gómez-Talegón, Inmaculada Fierro, M. Carmen Del Río, F. Javier Álvarez (UVa, University of Valladolid, Spain) Partners - Silvia Ravera, Susana Monteiro, Han de Gier (RUGPha, University of Groningen, the Netherlands) - Gertrude Van der Linden, Sara-Ann Legrand, Kristof Pil, Alain Verstraete (UGent, Ghent University, Belgium) - Michel Mallaret, Charles Mercier-Guyon, Isabelle Mercier-Guyon (UGren, University of Grenoble, Centre Regional de Pharmacovigilance, France) - Katerina Touliou (CERT-HIT, Centre for Research and Technology Hellas, Greece) - Michael Hei βing (BASt, Bundesanstalt für Straßenwesen, Germany). -

STOP TEST for PHARMACEUTICAL ANALYSIS 1 Point
Generated by Foxit PDF Creator © Foxit Software http://www.foxitsoftware.com For evaluation only. STOP TEST FOR PHARMACEUTICAL ANALYSIS 1 point 1. Indicator for direct aqueous titration is: a) potassium permanganate b) bromcresol green c) tropeolin OO d) phenophythaleine e) all answers are not correct 2. Indicator for direct non – aqueous titration of bases is: a) crystalviolet b) potassiun chromate c) methylorange d) methylene blue e) all answers are not correct 3. Titrant for direct non – aqueous titration of bases is: a) HCl b) AgNO3 c) H2SO4 d) HClO4 in CH3COOH e) all answers are not correct 4. Titrant for direct complexometric method is: a) NaNO2 b) KBrO 3 c) I2 d) KIO3 e) all answers are not correct 5. The name of direct argentometric titration is method of: a) Bouge – Lambert b) Schmidt – Kolbe c) Mor d) Traube e) all answers are not correct Generated by Foxit PDF Creator © Foxit Software http://www.foxitsoftware.com For evaluation only. 6. The name of indirect argentometric titration is method of: a) Folchard b) Hanch c) Kolbe – Schmidt d) Beer e) all answers are not correct 7. In titration with perchloric acid in glacials acetic acid to the determined hydrochoride of base is added: а) mercury acetate b) mercury chloride c) potassium bromide d) imidazole – mercury reactive e) all answers are not correct 8. In accordance to European Pharmacopoeia by direct cerimetry are determined the following substances: a) Paracetamol b) Ascorbic acid c) Nifedipine d) answers a), b), c) are correct e) answers a), b), c) are not correct 9. According to European Pharmacopoeia substances Menadione and Рaracеtamol are determined by: а) HPLC b) TLC c) spectrophotometry d) indirect cerimetry e) all answers are not correct 10. -

Principios Activos Fotosensibles
PRINCIPIOS ACTIVOS FOTOSENSIBLES PROTEGE A TUS PRODUCTOS DE LA FOTODEGRADACIÓN PRINCIPIOS ACTIVOS FOTOSENSIBLES Adenosine Carbamazepine Digitoxin Heptacaine Methaqualone-1-oxide Phenothiazines Sulfisomidine Tinidazole Vitamin D Adrenaline Carbisocaine Digoxin Hexachlorophane Methotrexate Phenylbutazone Sulpyrine Tolmetin Vitamin E Adriamycin Carboplatin Dihydroergotamin Hydralazine Metronidazole Phenylephrine Suprofen Tretinoin Vitamin K1 Amidopyrin Carmustine Dihydropyridines Mifepristone Phenytoin Triamcinolone Vitamin K2 Hydrochlorothiazide Suramin Amidinohydrazones Cefotaxime Diltiazem Hydrocortisone Minoxidil Physostigmine Triamterene Warfarin Amiloride Cefuroxime axetil Diosgenin Mitomycin C Piroxicam Tauromustine (TCNU) Trifluoperazine Hydroxychloroquine Terbutaline 4-Aminobenzoic acid Cephaeline Diphenhydramine Hypochloride Mitonafide Pralidoxime Trimethoprim Aminophenazone Cephalexine Dipyridamole Ibuprofen Mitoxantrone Prednisolone Tetracyclines Ubidecarenone Thiazide Aminophylline Cephradine Dithranol Imipramin Molsidomine Primaquine Vancomycin Thiocolchicoside Aminosalicylic acid Chalcone Dobutamin Indapamide Morphine Proguanil Vinblastine Thioridazine Amiodarone Chloramphenicol Dopamine Indomethacin Nalidixic acid Promazine Vitamin A Thiorphan Amodiaquine Chlordiazepoxide Dothiepin Indoprofen Naproxen Promethazine Vitamin B (see also Thiothixene Amonafide Chloroquine Doxorubicin Isoprenaline Neocarzinostatin Proxibarbital riboflavine) Tiaprofenic acid Amphotericin B Chlorpromazine DTIC Isopropylamino- Nimodipine Psoralen -

Drug and Medication Classification Schedule
KENTUCKY HORSE RACING COMMISSION UNIFORM DRUG, MEDICATION, AND SUBSTANCE CLASSIFICATION SCHEDULE KHRC 8-020-1 (11/2018) Class A drugs, medications, and substances are those (1) that have the highest potential to influence performance in the equine athlete, regardless of their approval by the United States Food and Drug Administration, or (2) that lack approval by the United States Food and Drug Administration but have pharmacologic effects similar to certain Class B drugs, medications, or substances that are approved by the United States Food and Drug Administration. Acecarbromal Bolasterone Cimaterol Divalproex Fluanisone Acetophenazine Boldione Citalopram Dixyrazine Fludiazepam Adinazolam Brimondine Cllibucaine Donepezil Flunitrazepam Alcuronium Bromazepam Clobazam Dopamine Fluopromazine Alfentanil Bromfenac Clocapramine Doxacurium Fluoresone Almotriptan Bromisovalum Clomethiazole Doxapram Fluoxetine Alphaprodine Bromocriptine Clomipramine Doxazosin Flupenthixol Alpidem Bromperidol Clonazepam Doxefazepam Flupirtine Alprazolam Brotizolam Clorazepate Doxepin Flurazepam Alprenolol Bufexamac Clormecaine Droperidol Fluspirilene Althesin Bupivacaine Clostebol Duloxetine Flutoprazepam Aminorex Buprenorphine Clothiapine Eletriptan Fluvoxamine Amisulpride Buspirone Clotiazepam Enalapril Formebolone Amitriptyline Bupropion Cloxazolam Enciprazine Fosinopril Amobarbital Butabartital Clozapine Endorphins Furzabol Amoxapine Butacaine Cobratoxin Enkephalins Galantamine Amperozide Butalbital Cocaine Ephedrine Gallamine Amphetamine Butanilicaine Codeine -

TEXAS RACING COMMISSION January 28, 2021 To
TEXAS RACING COMMISSION P. O. Box 12080, Austin, Texas 78711-2080 8505 Cross Park Drive, Suite 110, Austin, Texas 78754-4552 Phone (512) 833-6699 Fax (512) 833-6907 www.txrc.texas.gov January 28, 2021 To: Stewards, Commission Veterinarians, Test Barn Supervisors, Practicing Veterinarians, Owners, and Trainers From: Chuck Trout, Executive Director Re: Effective February 25, 2021 changes to the following documents: • Permissible Levels of Therapeutic Medications and Naturally Occurring Substances • Equine Medication Classification Policy and Penalty Guidelines • Equine Medication Classification List. This memo is to provide notice that the above listed documents are to be replaced effective this date. The changes include, but are not limited to: • Changes to the list of Permissible Levels of Therapeutic Medications and Naturally Occurring Substances; • Changes to the Equine Medication Classification Policy and Penalty Guidelines; • Changes to the Equine Medication Classification List. These documents are subject to further revision at any time. Test Barn Supervisors - please post this memo and the revised documents in the test barn as soon as possible. Also, please distribute copies of the Permissible Levels of Therapeutic Medications and Naturally Occurring Substances and Equine Medication Classification List to the practicing veterinarians at your racetrack. Licensing Staff - please post this memo and the revised documents where they may be viewed by the public as soon as possible. Copies of these documents will be made available on the Commission's website at http://www.txrc.texas.gov. Attachments: Permissible Levels of Therapeutic Medications and Naturally Occurring Substances Equine Medication Classification Policy and Penalty Guidelines Equine Medication Classification List TEXAS RACING COMMISSION P. -
Wo 2009/132050 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 29 October 2009 (29.10.2009) WO 2009/132050 A2 (51) International Patent Classification: 61/101,1 12 29 September 2008 (29.09.2008) US A61K 39/395 (2006.01) A61K 47/34 (2006.01) 61/140,033 22 December 2008 (22. 12.2008) US A61P 27/16 (2006.01) A61K 9/06 (2006.01) 61/160,233 13 March 2009 (13.03.2009) US (21) International Application Number: (71) Applicants (for all designated States except US): OTON- PCT/US2009/041320 OMY, INC. [US/US]; 5626 Oberlin Drive, Suite 100, San Diego, CA 92121 (US). THE REGENTS OF THE (22) International Filing Date: UNIVERSITY OF CALIFORNIA [US/US]; 1111 2 1 April 2009 (21 .04.2009) Franklin Street, 12th Floor, Oakland, CA 94607 (US). (25) Filing Language: English (72) Inventors; and (26) Publication Language: English (75) Inventors/Applicants (for US only): LICHTER, Jay [US/US]; P.O. Box 676244, Rancho Santa Fe, CA 92067 (30) Priority Data: (US). VOLLRATH, Benedikt [DE/US]; 4704 Niagara 61/046,543 2 1 April 2008 (21 .04.2008) US Avenue, San Diego, CA 92107 (US). TRAMMEL, An¬ 61/048,878 29 April 2008 (29.04.2008) US drew, M. [US/US]; 12485 South Alden Circle, Olathe, 61/127,7 13 14 May 2008 (14.05.2008) us KS 66062 (US). DURON, Sergio, G. [US/US]; 1605 61/055,625 23 May 2008 (23.05.2008) us Neale Street, San Diego, CA 92103 (US). -

ARCI Uniform Classification of Foreign Substances
DRUG TESTING STANDARDS AND PRACTICES PROGRAM. Uniform Classification Guidelines for Foreign Substances And Recommended Penalties Model Rule. September 2020 (V.14.2) © ASSOCIATION OF RACING COMMISSIONERS INTERNATIONAL – 2020. Association of Racing Commissioners International 2365 Harrodsburg Road, Suite B-450, Lexington, Kentucky USA 40504 www.arci.com Preamble to the Uniform Classification Guidelines of Foreign Substances The Preamble to the Uniform Classification Guidelines was approved by the RCI Drug Testing and Quality Assurance Program Committee (now the Drug Testing Standards and Practices Program Committee) on August 26, 1991. Minor revisions to the Preamble were made by the Drug Classification subcommittee (now the Veterinary Pharmacologists Subcommittee) on September 3, 1991. "The Uniform Classification Guidelines printed on the following pages are intended to assist stewards, hearing officers and racing commissioners in evaluating the seriousness of alleged violations of medication and prohibited substance rules in racing jurisdictions. Practicing equine veterinarians, state veterinarians, and equine pharmacologists are available and should be consulted to explain the pharmacological effects of the drugs listed in each class prior to any decisions with respect to penalties to be imposed. The ranking of drugs is based on their pharmacology, their ability to influence the outcome of a race, whether or not they have legitimate therapeutic uses in the racing horse, or other evidence that they may be used improperly. These classes of drugs are intended only as guidelines and should be employed only to assist persons adjudicating facts and opinions in understanding the seriousness of the alleged offenses. The facts of each case are always different and there may be mitigating circumstances which should always be considered. -

2021 Equine Prohibited Substances List
2021 Equine Prohibited Substances List . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). Prohibited Substances that are identified as Specified Substances in the List below should not in any way be considered less important or less dangerous than other Prohibited Substances. Rather, they are simply substances which are more likely to have been ingested by Horses for a purpose other than the enhancement of sport performance, for example, through a contaminated food substance. LISTED AS SUBSTANCE ACTIVITY BANNED 1-androsterone Anabolic BANNED 3β-Hydroxy-5α-androstan-17-one Anabolic BANNED 4-chlorometatandienone Anabolic BANNED 5α-Androst-2-ene-17one Anabolic BANNED 5α-Androstane-3α, 17α-diol Anabolic BANNED 5α-Androstane-3α, 17β-diol Anabolic BANNED 5α-Androstane-3β, 17α-diol Anabolic BANNED 5α-Androstane-3β, 17β-diol Anabolic BANNED 5β-Androstane-3α, 17β-diol Anabolic BANNED 7α-Hydroxy-DHEA Anabolic BANNED 7β-Hydroxy-DHEA Anabolic BANNED 7-Keto-DHEA Anabolic CONTROLLED 17-Alpha-Hydroxy Progesterone Hormone FEMALES BANNED 17-Alpha-Hydroxy Progesterone Anabolic MALES BANNED 19-Norandrosterone Anabolic BANNED 19-Noretiocholanolone Anabolic BANNED 20-Hydroxyecdysone Anabolic BANNED Δ1-Testosterone Anabolic BANNED Acebutolol Beta blocker BANNED Acefylline Bronchodilator BANNED Acemetacin Non-steroidal anti-inflammatory drug BANNED Acenocoumarol Anticoagulant CONTROLLED Acepromazine Sedative BANNED Acetanilid Analgesic/antipyretic CONTROLLED Acetazolamide Carbonic Anhydrase Inhibitor BANNED Acetohexamide Pancreatic stimulant CONTROLLED Acetominophen (Paracetamol) Analgesic BANNED Acetophenazine Antipsychotic BANNED Acetophenetidin (Phenacetin) Analgesic BANNED Acetylmorphine Narcotic BANNED Adinazolam Anxiolytic BANNED Adiphenine Antispasmodic BANNED Adrafinil Stimulant 1 December 2020, Lausanne, Switzerland 2021 Equine Prohibited Substances List . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). -

Drug/Substance Trade Name(S)
A B C D E F G H I J K 1 Drug/Substance Trade Name(s) Drug Class Existing Penalty Class Special Notation T1:Doping/Endangerment Level T2: Mismanagement Level Comments Methylenedioxypyrovalerone is a stimulant of the cathinone class which acts as a 3,4-methylenedioxypyprovaleroneMDPV, “bath salts” norepinephrine-dopamine reuptake inhibitor. It was first developed in the 1960s by a team at 1 A Yes A A 2 Boehringer Ingelheim. No 3 Alfentanil Alfenta Narcotic used to control pain and keep patients asleep during surgery. 1 A Yes A No A Aminoxafen, Aminorex is a weight loss stimulant drug. It was withdrawn from the market after it was found Aminorex Aminoxaphen, Apiquel, to cause pulmonary hypertension. 1 A Yes A A 4 McN-742, Menocil No Amphetamine is a potent central nervous system stimulant that is used in the treatment of Amphetamine Speed, Upper 1 A Yes A A 5 attention deficit hyperactivity disorder, narcolepsy, and obesity. No Anileridine is a synthetic analgesic drug and is a member of the piperidine class of analgesic Anileridine Leritine 1 A Yes A A 6 agents developed by Merck & Co. in the 1950s. No Dopamine promoter used to treat loss of muscle movement control caused by Parkinson's Apomorphine Apokyn, Ixense 1 A Yes A A 7 disease. No Recreational drug with euphoriant and stimulant properties. The effects produced by BZP are comparable to those produced by amphetamine. It is often claimed that BZP was originally Benzylpiperazine BZP 1 A Yes A A synthesized as a potential antihelminthic (anti-parasitic) agent for use in farm animals. -

Distinguishing Compounds with Anticancer Activity by ANN Using Inductive QSAR Descriptors
Bioinformation by Biomedical Informatics Publishing Group open access www.bioinformation.net Hypothesis ____________________________________________________________________________ Distinguishing compounds with anticancer activity by ANN using inductive QSAR descriptors Kunal Jaiswal1 and Pradeep Kumar Naik1, * 1Department of Biotechnology and Bioinformatics, Jaypee University of Information Technology, Waknaghat, Distt.-Solan, Himachal Pradesh, India-173215; Pradeep Kumar Naik* - E-mail: [email protected]; Phone: 91 1792 239227; * Corresponding author received May 19, 2008; revised June 17, 2008; accepted July 06, 2008; published July 30, 2008 Abstract: This article describes a method developed for predicting anticancer/non-anticancer drugs using artificial neural network (ANN). The ANN used in this study is a feed-forward neural network with a standard back-propagation training algorithm. Using 30 ‘inductive’ QSAR descriptors alone, we have been able to achieve 84.28% accuracy for correct separation of compounds with- and without anticancer activity. For the complete set of 30 inductive QSAR descriptors, ANN based method reveals a superior model (accuracy = 84.28%, Qpred = 74.28%, sensitivity = 0.9285, specificity = 0.7857, Matthews correlation coefficient (MCC) = 0.6998). The method was trained and tested on a non redundant data set of 380 drugs (122 anticancer and 258 non-anticancer). The elaborated QSAR model based on the Artificial Neural Networks approach has been extensively validated and has confidently assigned anticancer character to a number of trial anticancer drugs from the literature. Keywords: artificial neural network; inductive QSAR descriptors; anticancer drugs; non-anticancer drugs Background: A number of natural and synthetic products have been found Neural Networks (ANN) allow distinguishing biologically to exhibit anticancer activity against tumor cell lines [1, 2]. -

Uniform Classification Guidelines for Foreign Substances, Version 7.00
Association of Racing Commissioners International, Inc. Drug Testing Standards and Practices Program Model Rules Guidelines Uniform Classification Guidelines for Foreign Substances and Recommended Penalties and Model Rule Version 7.00 Revised January 2014 Association of Racing Commissioners International, Inc. Uniform Classification Guidelines for Foreign Substances Table of Contents Preamble to the Uniform Classification Guidelines of Foreign Substances ..................................................................................... ii Notes Regarding Classification Guidelines ...................................................................................................................................... ii Classification Criteria ....................................................................................................................................................................... iii Classification Definitions ................................................................................................................................................................. iv Drug Classification Scheme ............................................................................................................................................................. vi Alphabetical Substance List ............................................................................................................................................................... 1 Listing by Classification .................................................................................................................................................................. -
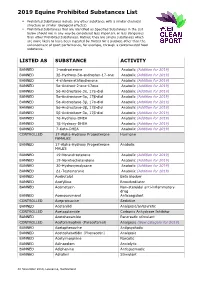
2019 Equine Prohibited Substances List
2019 Equine Prohibited Substances List . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). Prohibited Substances that are identified as Specified Substances in the List below should not in any way be considered less important or less dangerous than other Prohibited Substances. Rather, they are simply substances which are more likely to have been ingested by Horses for a purpose other than the enhancement of sport performance, for example, through a contaminated food substance. LISTED AS SUBSTANCE ACTIVITY BANNED 1-androsterone Anabolic (Addition for 2019) BANNED 3β-Hydroxy-5α-androstan-17-one Anabolic (Addition for 2019) BANNED 4-chlorometatandienone Anabolic (Addition for 2019) BANNED 5α-Androst-2-ene-17one Anabolic (Addition for 2019) BANNED 5α-Androstane-3α, 17α-diol Anabolic (Addition for 2019) BANNED 5α-Androstane-3α, 17β-diol Anabolic (Addition for 2019) BANNED 5α-Androstane-3β, 17α-diol Anabolic (Addition for 2019) BANNED 5α-Androstane-3β, 17β-diol Anabolic (Addition for 2019) BANNED 5β-Androstane-3α, 17β-diol Anabolic (Addition for 2019) BANNED 7α-Hydroxy-DHEA Anabolic (Addition for 2019) BANNED 7β-Hydroxy-DHEA Anabolic (Addition for 2019) BANNED 7-Keto-DHEA Anabolic (Addition for 2019) CONTROLLED 17-Alpha-Hydroxy Progesterone Hormone FEMALES BANNED 17-Alpha-Hydroxy Progesterone Anabolic MALES BANNED 19-Norandrosterone Anabolic (Addition for 2019) BANNED 19-Noretiocholanolone Anabolic (Addition for 2019) BANNED 20-Hydroxyecdysone Anabolic (Addition for 2019) BANNED