An in Vitro Model for Lysosomal Storage Diseases Using Aspartylglucosaminuria Patient Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fast Urinary Screening of Oligosaccharidoses by MALDI-TOF/TOF Mass Spectrometry
Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry. Laurent Bonesso, Monique Piraud, Céline Caruba, Emmanuel van Obberghen, Raymond Mengual, Charlotte Hinault To cite this version: Laurent Bonesso, Monique Piraud, Céline Caruba, Emmanuel van Obberghen, Raymond Mengual, et al.. Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry.. Orphanet Journal of Rare Diseases, BioMed Central, 2014, 9 (1), pp.19. 10.1186/1750-1172-9-19. inserm- 00945684 HAL Id: inserm-00945684 https://www.hal.inserm.fr/inserm-00945684 Submitted on 12 Feb 2014 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Bonesso et al. Orphanet Journal of Rare Diseases 2014, 9:19 http://www.ojrd.com/content/9/1/19 RESEARCH Open Access Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry Laurent Bonesso1, Monique Piraud5, Céline Caruba1, Emmanuel Van Obberghen1,2,3,4, Raymond Mengual1† and Charlotte Hinault1,2,3,4*† Abstract Background: Oligosaccharidoses, which belong to the lysosomal storage diseases, are inherited metabolic disorders due to the absence or the loss of function of one of the enzymes involved in the catabolic pathway of glycoproteins and indirectly of glycosphingolipids. -

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -

Oligosaccharide Screen, Urine
Lab Dept: Urine/Stool Test Name: OLIGOSACCHARIDE SCREEN, URINE General Information Lab Order Codes: OLGO Synonyms: N/A CPT Codes: 84376 – Sugars (mono-, di-, oligosaccharides); single qualitative, each specimen Test Includes: This is a screening method for a subset of lysosomal storage disorders including: alpha-mannosidosis, aspartylglucosaminuria, fucosidosis, Schindler disease, GM1 gangliosidosis, Sandhoff disease, sialidosis, galactosialidosis, mucolipidoses types II and III, and Pompe disease. Logistics Test Indications: Investigation of possible oligosaccharidoses Lab Testing Sections: Chemistry - Sendouts Referred to: Mayo Medical Laboratories (MML Test: OLIGU) Phone Numbers: MIN Lab: 612-813-6280 STP Lab: 651-220-6550 Test Availability: Daily, 24 hours Turnaround Time: 4 – 8 days; performed Monday and Wednesday Special Instructions: Include family history, clinical condition (asymptomatic or acute episode), diet, and drug therapy information. Specimen Specimen Type: Urine, random collection Container: Plastic leakproof container (No preservatives) Draw Volume: 8 mL (Minimum: 2 mL) urine Processed Volume: Same as Draw Volume Collection: A random urine sample may be obtained by voiding into a urine cup and is often performed at the laboratory. Bring the refrigerated container to the lab. Make sure all specimens submitted to the laboratory are properly labeled with the patient’s name, medical record number and date of birth. Special Processing: Lab Staff: Aliquot specimen into a 13 mL urine tube, no preservative. Freeze immediately. Store and ship at frozen temperatures. Patient Preparation: None Sample Rejection: Mislabeled or unlabeled specimens Interpretive Reference Range: An interpretive report will be provided. This is a screening test; not all oligosachharidoses are detected. The resulting excretion profile may be characteristic of a specific disorder; however, abnormal results require confirmation by enzyme assay or molecular genetic testing. -

Allo HCT for Metabolic Disorders and Severe Osteopetrosis Status: Recruiting
MT2013-31: Allo HCT for Metabolic Disorders and Severe Osteopetrosis Status: Recruiting Eligibility Criteria Sex: All Age: up to 55 Years old This study is NOT accepting healthy volunteers Inclusion Criteria: • 0 through 55 years of age • Adequate graft available • Adequate organ function • Eligible Diseases: • Mucopolysaccharidosis Disorders: • MPS IH (Hurler syndrome) • MPS II (Hunter syndrome) if the patient has no or minimal evidence of symptomatic neurologic disease but is expected to have a neurologic phenotype • MPS VI (Maroteaux-Lamy syndrome) • MPS VII (Sly syndrome) • Glycoprotein Metabolic Disorders: • Alpha mannosidosis • Fucosidosis • Aspartylglucosaminuria • Sphingolipidoses and Recessive Leukodystrophies: • Globoid cell leukodystrophy • Metachromatic leukodystrophy • Niemann-Pick B patients (sphingomyelin deficiency) • Niemann-Pick C subtype 2 • Peroxisomal Disorders: • Adrenoleukodystrophy with cerebral involvement • Zellweger syndrome • Neonatal Adrenoleukodystrophy • Infantile Refsum disease • Acyl-CoA-Oxidase Deficiency • D-Bifunctional enzyme deficiency • Multifunctional enzyme deficiency • Alpha-methylacyl-CoA Racmase Deficiency (AMACRD) • Mitochondrial Neurogastrointestingal Encephalopathy (MNGIE) • Severe Osteopetrosis (OP) • Hereditary Leukoencephalopathy with axonal spheroids (HDLS; CSF1R mutation) • Other Inherited Metabolic Disorders (IMD): Patients will also be considered who have other life-threatening, rare lysosomal, peroxisomal or other similar inherited disorders characterized by white matter disease or -

A Case Report
Open Access Case Report DOI: 10.7759/cureus.6139 Diagnosis and Supportive Management of Fucosidosis: A Case Report Arpanjeet Kaur 1 , Arshdeep S. Dhaliwal 2 , Hillary Raynes 3 , Thomas P. Naidich 4 , David M. Kaufman 3 1. General Medicine, Sibia Healthcare Pvt. Ltd., Sangrur, IND 2. General Medicine, Madhu Nursing Home, Patiala, IND 3. Pediatric Neurology, Mount Sinai Hospital, New York, USA 4. Radiology, Mount Sinai Hospital, New York, USA Corresponding author: Arshdeep S. Dhaliwal, [email protected] Abstract Fucosidosis is a lysosomal storage disease, resulting from a deficiency of the enzyme alpha-L-fucosidase. We present the case of an affected female with numerous manifestations, clinically and radiographically. In fucosidosis, advanced interventions are not always necessary to have rewarding outcomes. In fact, early diagnosis and management of the symptoms with a multi-systemic supportive care approach can improve the quality of life and may also prolong the life of those patients diagnosed with fucosidosis. Categories: Genetics, Neurology, Pediatrics Keywords: diagnosis, fucosidosis, fuca1, supportive management Introduction Fucosidosis is a very rare, autosomal, recessive genetic disorder associated with a mutation in the FUCA1 gene, resulting in a severe deficiency of the enzyme alpha-L-fucosidase in lysosomes. A total of 100 cases of fucosidosis have been reported until now [1-2]. Deficiency of this enzyme leads to the accumulation of fucose-containing glycolipids and other substances in various organs, which display a wide array of symptoms, such as severe global developmental delay (95%), muscle stiffness (87%), coarse facies (79%), recurrent respiratory infections (78%), abnormal bone development (58%), tortuous conjunctival vessels (53%), clusters of enlarged blood vessels on the skin (52%), abnormal enlargement of visceral organs (44%), and seizures (38%) [3-6]. -

Human Xc-N-Acetylgalactosaminidase Qx-NAGA)
4585 JMed Genet 1996;33:458-464 Human xc-N-acetylgalactosaminidase Qx-NAGA) deficiency: new mutations and the paradox J Med Genet: first published as 10.1136/jmg.33.6.458 on 1 June 1996. Downloaded from between genotype and phenotype J L M Keulemans, A J J Reuser, M A Kroos, R Willemsen, M M P Hermans, A M W van den Ouweland, J G N de Jong, R A Wevers, W 0 Renier, D Schindler, M J Coll, A Chabas, H Sakuraba, Y Suzuki, 0 P van Diggelen Abstract reported the second independent case of a- Up to now eight patients with a-NAGA NAGA deficiency with an entirely different deficiency have been described. This in- clinical phenotype. This patient had a late onset cludes the newly identified patient re- disease with slight facial coarseness, dis- ported here who died unexpectedly aged seminated angiokeratoma, and mild intellectual II years of hypoxia during convulsions; impairment (IQ= 70), but without neuro- necropsy was not performed. logical symptoms. Unlike the infantile cases, Three patients have been genotyped pre- this patient had prominent vacuolisation in all viously and here we report the mutations dermal cells, most prominently in vascular and in the other five patients, including two lymphatic endothelial cells and eccrine sweat new mutations (S160C and E193X). The gland cells, but also in dermal neural cells and newly identified patient is consanguineous fibroblasts.' The glomerular endothelial cells with the first patients reported with a- but not the epithelial kidney cells are involved NAGA deficiency and neuroaxonal dys- and also blood lymphocytes are vacuolised.6 trophy and they all had the a-NAGA geno- These three patients shared, however, the type E325KIE325K. -

The Myriad Foresight® Carrier Screen
The Myriad Foresight® Carrier Screen 180 Kimball Way | South San Francisco, CA 94080 www.myriadwomenshealth.com | [email protected] | (888) 268-6795 The Myriad Foresight® Carrier Screen - Disease Reference Book 11-beta-hydroxylase-deficient Congenital Adrenal Hyperplasia ...............................................................................................................................................................................8 6-pyruvoyl-tetrahydropterin Synthase Deficiency....................................................................................................................................................................................................10 ABCC8-related Familial Hyperinsulinism..................................................................................................................................................................................................................12 Adenosine Deaminase Deficiency ............................................................................................................................................................................................................................14 Alpha Thalassemia ....................................................................................................................................................................................................................................................16 Alpha-mannosidosis ..................................................................................................................................................................................................................................................18 -

SSIEM Classification of Inborn Errors of Metabolism 2011
SSIEM classification of Inborn Errors of Metabolism 2011 Disease group / disease ICD10 OMIM 1. Disorders of amino acid and peptide metabolism 1.1. Urea cycle disorders and inherited hyperammonaemias 1.1.1. Carbamoylphosphate synthetase I deficiency 237300 1.1.2. N-Acetylglutamate synthetase deficiency 237310 1.1.3. Ornithine transcarbamylase deficiency 311250 S Ornithine carbamoyltransferase deficiency 1.1.4. Citrullinaemia type1 215700 S Argininosuccinate synthetase deficiency 1.1.5. Argininosuccinic aciduria 207900 S Argininosuccinate lyase deficiency 1.1.6. Argininaemia 207800 S Arginase I deficiency 1.1.7. HHH syndrome 238970 S Hyperammonaemia-hyperornithinaemia-homocitrullinuria syndrome S Mitochondrial ornithine transporter (ORNT1) deficiency 1.1.8. Citrullinemia Type 2 603859 S Aspartate glutamate carrier deficiency ( SLC25A13) S Citrin deficiency 1.1.9. Hyperinsulinemic hypoglycemia and hyperammonemia caused by 138130 activating mutations in the GLUD1 gene 1.1.10. Other disorders of the urea cycle 238970 1.1.11. Unspecified hyperammonaemia 238970 1.2. Organic acidurias 1.2.1. Glutaric aciduria 1.2.1.1. Glutaric aciduria type I 231670 S Glutaryl-CoA dehydrogenase deficiency 1.2.1.2. Glutaric aciduria type III 231690 1.2.2. Propionic aciduria E711 232000 S Propionyl-CoA-Carboxylase deficiency 1.2.3. Methylmalonic aciduria E711 251000 1.2.3.1. Methylmalonyl-CoA mutase deficiency 1.2.3.2. Methylmalonyl-CoA epimerase deficiency 251120 1.2.3.3. Methylmalonic aciduria, unspecified 1.2.4. Isovaleric aciduria E711 243500 S Isovaleryl-CoA dehydrogenase deficiency 1.2.5. Methylcrotonylglycinuria E744 210200 S Methylcrotonyl-CoA carboxylase deficiency 1.2.6. Methylglutaconic aciduria E712 250950 1.2.6.1. Methylglutaconic aciduria type I E712 250950 S 3-Methylglutaconyl-CoA hydratase deficiency 1.2.6.2. -

Collectively Common, Individually Rare Many
OUTLOOK LYSOSOMAL STORAGE DISORDERS MYRIAD MALADIES u A tophag os om e Mutations affecting an es enzyme that labels other som do 3 enzymes for delivery to n the lysosome result in E 4 mucolipidosis-II Defects in a protein that helps endosomes and autophagosomes fuse with the lysosome cause Danon disease Defects in processing enzymes can cause Ly undigested material to Go soso lgi 2 me accumulate, such as in Gaucher’s disease and 5 Hunter syndrome En doplas mic retic ulum 1 Failure of processing in the endoplasmic reticulum can inac- 6 tivate a whole class Loss of transport proteins of enzymes, such as causes the lysosome to retain molecular building blocks in Nucleus in multiple sulfatase deficiency diseases such as cystinosis or sialic acid storage disease MANY MOVING PARTS The lysosome uses specialized enzymes to help cells to digest external biological materials and recycle defective proteins and damaged cellular machinery. Many LSDs arise from mutations in the genes that encode those enzymes, but there are numerous other ways in which this process can break down (red stars). 1 The endoplasmic The Golgi apparatus Endosomes transport The autophagosome Within the lysosome, Molecular building blocks reticulum performs the further processes and enzymes from the Golgi delivers damaged enzymes convert are released into cell for initial processing of labels specific enzymes and materials from organelles and misfolded molecules such as sugars, reuse newly synthesized for delivery to the outside the cell to the proteins to the lysosome proteins and lipids into lysosomal enzymes lysosome lysosome for recycling simpler building blocks COLLECTIVELY COMMON, INDIVIDUALLY RARE LSDs are not especially rare; estimates suggest that 1 in just over 5,000 newborns newborns worldwide, and the rarest have been described only a handful of times. -

Conventional and Unconventional Therapeutic Strategies for Sialidosis Type I
Journal of Clinical Medicine Article Conventional and Unconventional Therapeutic Strategies for Sialidosis Type I 1, 1, 1 1,2 Rosario Mosca y, Diantha van de Vlekkert y , Yvan Campos , Leigh E. Fremuth , Jaclyn Cadaoas 3 , Vish Koppaka 3, Emil Kakkis 3, Cynthia Tifft 4, Camilo Toro 5 , Simona Allievi 6,7, Cinzia Gellera 6,7, Laura Canafoglia 7 , Gepke Visser 8 , Ida Annunziata 1 and Alessandra d’Azzo 1,* 1 Department of Genetics, St. Jude Children’s Research Hospital, Memphis, TN 38105, USA; [email protected] (R.M.); [email protected] (D.v.d.V.); [email protected] (Y.C.); [email protected] (L.E.F.); [email protected] (I.A.) 2 Department of Anatomy and Neurobiology, College of Graduate Health Sciences, University of Tennessee Health Science Center, Memphis, TN 38163, USA 3 Ultragenyx Pharmaceutical, Novato, CA 94949, USA; [email protected] (J.C.); [email protected] (V.K.); [email protected] (E.K.) 4 Office of the Clinical Director & Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health (NHGRI), Bethesda, MD 20892, USA; [email protected] 5 Undiagnosed Disease Network, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA; [email protected] 6 Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milan, Italy; [email protected] (S.A.); [email protected] (C.G.) 7 Neurophysiopathology, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milan, Italy; [email protected] 8 Department of Metabolic Diseases, Wilhelmina Children’s Hospital, University Medical Center Utrecht, 3584 CX Utrecht, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +1-901-595-2698 These authors contributed equally to this work. -
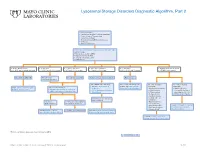
Lysosomal Storage Disorders Diagnostic Algorithm, Part 2
Lysosomal Storage Disorders Diagnostic Algorithm, Part 2 Clinical information: ■ Developmental delay/Cognitive impairment ■ Coarse features/Organomegaly ■ Dysostosis multiplex ■ Neurodegeneration/Behavioral changes ■ Ichthyosis ■ Hearing defects/loss LSDS / Lysosomal Storage Disorders Screen, Random, Urine Testing includes: ■ Mucopolysaccharides (MPS) ■ Oligosaccharides (OLIGO) ■ Ceramide trihexosides (CT) ■ Sulfatides (S) ■ OLIGO: ML II/III profile ■ S: abnormal ■ CT and S: abnormal ■ MPS and S: abnormal ■ CT: abnormal ■ OLIGO: characteristic profile ■ CT, MPS and S: normal/abnormal ■ CT, MPS and OLIGO: normal ■ MPS and OLIGO: normal ■ CT and OLIGO: normal ■ MPS, OLIGO and S: normal ■ CT, MPS and S; normal Mucolipidosis (ML) II/III Metachromatic Prosaposin/ SaposinB Multiple sulfatase deficiency (MSD) Fabry disease leukodystrophy (MLD) Order BOTH of the following: For recommended diagnostic One of the following One of the following ■ ARSAW / Arylsulfatase A, workup, see Fabry Disease suspected: suspected: Order: GNPTZ / GNPTAB Gene, Order 1 of the following: Leukocytes Diagnostic Testing Algorithm ■ Aspartylglucosaminuria ■ NGLY1 deficiency Full Gene Analysis, Varies ■ ARSAW / Arylsulfatase A, Leukocytes ■ I2SW / Iduronate-2-Sulfatase, ■ α-Mannosidosis (Congenital disorder of ■ ARSU / Arylsulfatase A, 24 Hour, Urine Whole Blood ■ β-Mannosidosis glycosylation: CDG-Iv) ■ Pompe disease ■ MOGS-CDG (CDG-IIb) ■ Sandhoff disease ■ Schindler disease If both results are deficient, ■ Sialidosis MSD confirmed If deficient, If normal, possible Saposin -

Sialidosis Type I Presenting with a Novel Mutation and Advanced Neuroimaging Features
Case Reports Sialidosis type I presenting with a novel mutation and advanced neuroimaging features Murat Gultekin, MD, Ruslan Bayramov, MD, Cagatay Karaca, MD, Niyazi Acer, PhD. ABSTRACT Received 16th July 2017. Accepted 29th November 2017. Address correspondence and reprint request to: Dr. Murat Gultekin, ,Department of Neurology, Erciyes University School of Medicine داء اللعاب هو مرض تخزين نورامينيداز النادر والناجمة عن طفرة Kayseri, Turkey. E-mail: [email protected] جينات نيورامينيداز (NEU1) ونقص في أنزمي نورامينيداز. وكان الهدف من هذه الدراسة هو دراسة نوع داء اللعاب الدماغي النوع األول باستخدام التصوير بالرنني املغناطيسي )MRI(، التصوير #ialidosis )Neuraminidase deficiency, OMIM االنتشاري للعضلة والتصوير بالرنني املغناطيسي الوظيفي باملقارنة S256550( is a rare autosomal recessive disease with مع 3 التحكم. وحدد حتليل اجلني للمريض طفرة متغايرة مركب approximate prevalence of 1/5,000,000-1/1,500,000 في NEU1 الذي يظهر أنه يرتبط مع نوع داء اللعاب 1. في (live births, related to neuraminidase gene (NEU1 هذه احلالة نادرا جدا ، وجدنا تغيرات في حجم الهياكل الدماغ )OMIM * 608272( pathogenic variants, leading to 1 املختلفة. وجدنا أن أحجام النواة حتت املهاد أصغر في املريض 1 neuraminidase )EC 3.2.1.18( deficiency. Due to age ;of onset and severity, Sialidosis divided into 2 types مقارنة مع التحكم. أيضا، كان داء اللعاب أصغر حجم املخيخ milder, late-onset, non-dysmorphic form, characterized مقارنة مع مجموعة السيطرة بشكل ملحوظ. وكانت احلالة أعلى ,by macular cherry-red spot, visual defects, myoclonus معدل االنتشار ونسبة قيم أقل لتفاوت االنكسار في املخيخ وظهر -ataxia, and seizures - Sialidosis type 1 and severe, early اتصال وظيفي غير طبيعي. onset, dysmorphic form - Sialidosis type 2.2 Myoclonic Sialidosis is a rare lysosomal storage disease caused movements are known to have many possible causes.