Lecture 5 — Scattering Geometries
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Classical and Modern Diffraction Theory
Downloaded from http://pubs.geoscienceworld.org/books/book/chapter-pdf/3701993/frontmatter.pdf by guest on 29 September 2021 Classical and Modern Diffraction Theory Edited by Kamill Klem-Musatov Henning C. Hoeber Tijmen Jan Moser Michael A. Pelissier SEG Geophysics Reprint Series No. 29 Sergey Fomel, managing editor Evgeny Landa, volume editor Downloaded from http://pubs.geoscienceworld.org/books/book/chapter-pdf/3701993/frontmatter.pdf by guest on 29 September 2021 Society of Exploration Geophysicists 8801 S. Yale, Ste. 500 Tulsa, OK 74137-3575 U.S.A. # 2016 by Society of Exploration Geophysicists All rights reserved. This book or parts hereof may not be reproduced in any form without permission in writing from the publisher. Published 2016 Printed in the United States of America ISBN 978-1-931830-00-6 (Series) ISBN 978-1-56080-322-5 (Volume) Library of Congress Control Number: 2015951229 Downloaded from http://pubs.geoscienceworld.org/books/book/chapter-pdf/3701993/frontmatter.pdf by guest on 29 September 2021 Dedication We dedicate this volume to the memory Dr. Kamill Klem-Musatov. In reading this volume, you will find that the history of diffraction We worked with Kamill over a period of several years to compile theory was filled with many controversies and feuds as new theories this volume. This volume was virtually ready for publication when came to displace or revise previous ones. Kamill Klem-Musatov’s Kamill passed away. He is greatly missed. new theory also met opposition; he paid a great personal price in Kamill’s role in Classical and Modern Diffraction Theory goes putting forth his theory for the seismic diffraction forward problem. -

Neutron Diffraction Patterns Measured with a High-Resolution Powder Diffractometer Installed on a Low-Flux Reactor
Neutron diffraction patterns measured with a high-resolution powder diffractometer installed on a low-flux reactor V. L. Mazzocchi1, C. B. R. Parente1, J. Mestnik-Filho1 and Y. P. Mascarenhas2 1 Instituto de Pesquisas Energéticas e Nucleares (IPEN-CNEN/SP), CP 11049, 05422-970, São Paulo, SP, Brazil 2 Instituto de Física de São Carlos (IFSCAR-USP), CP 369, 13560-940, São Carlos, SP, Brazil E-mail of corresponding author: [email protected] International Conference on Research Reactors 14-18 November 2011 Rabat, Morocco Research Reactor Center (CRPq) CRPq’s main program - Nuclear and condensed matter physics - Neutron activation analysis - Nuclear metrology - Applied nuclear physics - Graduate and postgraduate teaching - Reactor operators training IEA-R1 Reactor - Swimming pool type, light water moderated with 23 graphite and 9 beryllium reflectors, designed to operate at 5 MW - Current power: 4.5 MW - Neutron in core flux: 7,0 x 1013n/cm2.s - Suitable for the use in: basic and applied research, production of medical radioisotopes, industry and natural sciences applications. International Conference on Research Reactors 14-18 November 2011 Rabat, Morocco 1- Characteristics of the old and the new neutron diffractometers The old IPEN-CNEN/SP neutron multipurpose diffractometer . a single BF3 detector . a single wavelength ( =1.137 Å) . a point-to-point scanning data measurement The new IPEN-CNEN/SP neutron powder diffractometer . a Position Sensitive Detector (PSD) array formed by 11 linear proportional 3He detectors, scanning a 2 = 20° interval -

Intensity of Double-Slit Pattern • Three Or More Slits
• Intensity of double-slit pattern • Three or more slits Practice: Chapter 37, problems 13, 14, 17, 19, 21 Screen at ∞ θ d Path difference Δr = d sin θ zero intensity at d sinθ = (m ± ½ ) λ, m = 0, ± 1, ± 2, … max. intensity at d sinθ = m λ, m = 0, ± 1, ± 2, … 1 Questions: -what is the intensity of the double-slit pattern at an arbitrary position? -What about three slits? four? five? Find the intensity of the double-slit interference pattern as a function of position on the screen. Two waves: E1 = E0 sin(ωt) E2 = E0 sin(ωt + φ) where φ =2π (d sin θ )/λ , and θ gives the position. Steps: Find resultant amplitude, ER ; then intensities obey where I0 is the intensity of each individual wave. 2 Trigonometry: sin a + sin b = 2 cos [(a-b)/2] sin [(a+b)/2] E1 + E2 = E0 [sin(ωt) + sin(ωt + φ)] = 2 E0 cos (φ / 2) cos (ωt + φ / 2)] Resultant amplitude ER = 2 E0 cos (φ / 2) Resultant intensity, (a function of position θ on the screen) I position - fringes are wide, with fuzzy edges - equally spaced (in sin θ) - equal brightness (but we have ignored diffraction) 3 With only one slit open, the intensity in the centre of the screen is 100 W/m 2. With both (identical) slits open together, the intensity at locations of constructive interference will be a) zero b) 100 W/m2 c) 200 W/m2 d) 400 W/m2 How does this compare with shining two laser pointers on the same spot? θ θ sin d = d Δr1 d θ sin d = 2d θ Δr2 sin = 3d Δr3 Total, 4 The total field is where φ = 2π (d sinθ )/λ •When d sinθ = 0, λ , 2λ, .. -

Resolution Neutron Scattering Technique Using Triple-Axis
1 High -resolution neutron scattering technique using triple-axis spectrometers ¢ ¡ ¡ GUANGYONG XU, ¡ * P. M. GEHRING, V. J. GHOSH AND G. SHIRANE ¢ ¡ Physics Department, Brookhaven National Laboratory, Upton, NY 11973, and NCNR, National Institute of Standards and Technology, Gaithersburg, Maryland, 20899. E-mail: [email protected] (Received 0 XXXXXXX 0000; accepted 0 XXXXXXX 0000) Abstract We present a new technique which brings a substantial increase of the wave-vector £ - resolution of triple-axis-spectrometers by matching the measurement wave-vector £ to the ¥§¦©¨ reflection ¤ of a perfect crystal analyzer. A relative Bragg width of can be ¡ achieved with reasonable collimation settings. This technique is very useful in measuring small structural changes and line broadenings that can not be accurately measured with con- ventional set-ups, while still keeping all the strengths of a triple-axis-spectrometer. 1. Introduction Triple-axis-spectrometers (TAS) are widely used in both elastic and inelastic neutron scat- tering measurements to study the structures and dynamics in condensed matter. It has the £ flexibility to allow one to probe nearly all coordinates in energy ( ) and momentum ( ) space in a controlled manner, and the data can be easily interpreted (Bacon, 1975; Shirane et al., 2002). The resolution of a triple-axis-spectrometer is determined by many factors, including the ¨ incident (E ) and final (E ) neutron energies, the wave-vector transfer , the monochromator PREPRINT: Acta Crystallographica Section A A Journal of the International Union of Crystallography 2 and analyzer mosaic, and the beam collimations, etc. This has been studied in detail by Cooper & Nathans (1967), Werner & Pynn (1971) and Chesser & Axe (1973). -

Design Guidelines for an Electron Diffractometer for Structural Chemistry and Structural Biology
research papers Design guidelines for an electron diffractometer for structural chemistry and structural biology ISSN 2059-7983 Jonas Heidler,a Radosav Pantelic,b Julian T. C. Wennmacher,a Christian Zaubitzer,c Ariane Fecteau-Lefebvre,d Kenneth N. Goldie,d Elisabeth Mu¨ller,a Julian J. Holstein,e Eric van Genderen,a Sacha De Carlob and Tim Gruenea*‡ Received 20 December 2018 aPaul Scherrer Institut, 5232 Villigen PSI, Switzerland, bDECTRIS Ltd, 5405 Baden-Daettwil, Switzerland, cScientific Accepted 22 March 2019 Center for Optical and Electron Microscopy, ETH Zu¨rich, 8093 Zu¨rich, Switzerland, dCenter for Cellular Imaging and NanoAnalytics, University Basel, 4058 Basel, Switzerland, and eFaculty of Chemistry and Chemical Biology, TU Dortmund University, Otto Hahn Strasse 6, 44227 Dortmund, Germany. *Correspondence e-mail: [email protected] ‡ Current address: Zentrum fu¨rRo¨ntgen- strukturanalyse, Faculty of Chemistry, Universita¨t Wien, 1090 Wien, Austria. 3D electron diffraction has reached a stage where the structures of chemical compounds can be solved productively. Instrumentation is lagging behind this Keywords: electron diffractometer; EIGER X 1M development, and to date dedicated electron diffractometers for data collection detector; 3D electron diffraction; chemical crystallography; EIGER hybrid pixel detector; based on the rotation method do not exist. Current studies use transmission structural chemistry. electron microscopes as a workaround. These are optimized for imaging, which is not optimal for diffraction studies. The beam intensity is very high, it is difficult to create parallel beam illumination and the detectors used for imaging are of only limited use for diffraction studies. In this work, the combination of an EIGER hybrid pixel detector with a transmission electron microscope to construct a productive electron diffractometer is described. -

Development of an X Ray Diffractometer and Its Safety Features
DEVELOPMENT OF AN X RAY DIFFRACTOMETER AND ITS SAFETY FEATURES By Jordan A. Cady A thesis submitted in partial fulfillment of the requirements for the degree of Bachelor of Science Houghton College January 2017 Signature of Author…………………………………………….…………………….. Department of Physics January 4, 2017 …………………………………………………………………………………….. Dr. Brandon Hoffman Associate Professor of Physics Research Supervisor …………………………………………………………………………………….. Dr. Mark Yuly Professor of Physics DEVELOPMENT OF AN X RAY DIFFRACTOMETER AND ITS SAFETY FEATURES By Jordan A. Cady Submitted to the Department of Physics on January 4, 2017 in partial fulfillment of the requirement for the degree of Bachelor of Science Abstract A Braggs-Brentano θ-2θ x ray diffractometer is being constructed at Houghton College to map the microstructure of textured, polycrystalline silver films. A Phillips-Norelco x ray source will be used in conjunction with a 40 kV power supply. The motors for the motion of the θ and 2θ arms, as well as a Vernier Student Radiation Monitor, will all be controlled by a program written in LabVIEW. The entire mechanical system and x ray source are contained in a steel enclosure to ensure the safety of the users. Thesis Supervisor: Dr. Brandon Hoffman Title: Associate Professor of Physics 2 TABLE OF CONTENTS Chapter 1 Introduction ...........................................................................................6 1.1 History of Interference ............................................................................6 1.2 Discovery of X rays .................................................................................7 -

Light and Matter Diffraction from the Unified Viewpoint of Feynman's
European J of Physics Education Volume 8 Issue 2 1309-7202 Arlego & Fanaro Light and Matter Diffraction from the Unified Viewpoint of Feynman’s Sum of All Paths Marcelo Arlego* Maria de los Angeles Fanaro** Universidad Nacional del Centro de la Provincia de Buenos Aires CONICET, Argentine *[email protected] **[email protected] (Received: 05.04.2018, Accepted: 22.05.2017) Abstract In this work, we present a pedagogical strategy to describe the diffraction phenomenon based on a didactic adaptation of the Feynman’s path integrals method, which uses only high school mathematics. The advantage of our approach is that it allows to describe the diffraction in a fully quantum context, where superposition and probabilistic aspects emerge naturally. Our method is based on a time-independent formulation, which allows modelling the phenomenon in geometric terms and trajectories in real space, which is an advantage from the didactic point of view. A distinctive aspect of our work is the description of the series of transformations and didactic transpositions of the fundamental equations that give rise to a common quantum framework for light and matter. This is something that is usually masked by the common use, and that to our knowledge has not been emphasized enough in a unified way. Finally, the role of the superposition of non-classical paths and their didactic potential are briefly mentioned. Keywords: quantum mechanics, light and matter diffraction, Feynman’s Sum of all Paths, high education INTRODUCTION This work promotes the teaching of quantum mechanics at the basic level of secondary school, where the students have not the necessary mathematics to deal with canonical models that uses Schrodinger equation. -
Structure Factors Again
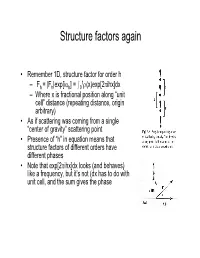
Structure factors again • Remember 1D, structure factor for order h 1 –Fh = |Fh|exp[iαh] = I0 ρ(x)exp[2πihx]dx – Where x is fractional position along “unit cell” distance (repeating distance, origin arbitrary) • As if scattering was coming from a single “center of gravity” scattering point • Presence of “h” in equation means that structure factors of different orders have different phases • Note that exp[2πihx]dx looks (and behaves) like a frequency, but it’s not (dx has to do with unit cell, and the sum gives the phase Back and Forth • Fourier sez – For any function f(x), there is a “transform” of it which is –F(h) = Ûf(x)exp(2pi(hx))dx – Where h is reciprocal of x (1/x) – Structure factors look like that • And it works backward –f(x) = ÛF(h)exp(-2pi(hx))dh – Or, if h comes only in discrete points –f(x) = SF(h)exp(-2pi(hx)) Structure factors, cont'd • Structure factors are a "Fourier transform" - a sum of components • Fourier transforms are reversible – From summing distribution of ρ(x), get hth order of diffraction – From summing hth orders of diffraction, get back ρ(x) = Σ Fh exp[-2πihx] Two dimensional scattering • In Frauenhofer diffraction (1D), we considered scattering from points, along the line • In 2D diffraction, scattering would occur from lines. • Numbering of the lines by where they cut the edges of a unit cell • Atom density in various lines can differ • Reflections now from planes Extension to 3D – Planes defined by extension from 2D case – Unit cells differ • Depends on arrangement of materials in 3D lattice • = "Space -

System Design and Verification of the Precession Electron Diffraction Technique
NORTHWESTERN UNIVERSITY System Design and Verification of the Precession Electron Diffraction Technique A DISSERTATION SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILLMENT OF THE REQUIREMENTS for the degree DOCTOR OF PHILOSOPHY Field of Materials Science and Engineering By Christopher Su-Yan Own EVANSTON, ILLINOIS First published on the WWW 01, August 2005 Build 05.12.07. PDF available for download at: http://www.numis.northwestern.edu/Research/Current/precession.shtml c Copyright by Christopher Su-Yan Own 2005 All Rights Reserved ii ABSTRACT System Design and Verification of the Precession Electron Diffraction Technique Christopher Su-Yan Own Bulk structural crystallography is generally a two-part process wherein a rough starting structure model is first derived, then later refined to give an accurate model of the structure. The critical step is the deter- mination of the initial model. As materials problems decrease in length scale, the electron microscope has proven to be a versatile and effective tool for studying many problems. However, study of complex bulk structures by electron diffraction has been hindered by the problem of dynamical diffraction. This phenomenon makes bulk electron diffraction very sensitive to specimen thickness, and expensive equip- ment such as aberration-corrected scanning transmission microscopes or elaborate methodology such as high resolution imaging combined with diffraction and simulation are often required to generate good starting structures. The precession electron diffraction technique (PED), which has the ability to significantly reduce dynamical effects in diffraction patterns, has shown promise as being a “philosopher’s stone” for bulk electron diffraction. However, a comprehensive understanding of its abilities and limitations is necessary before it can be put into widespread use as a standalone technique. -

Form and Structure Factors: Modeling and Interactions Jan Skov Pedersen, Department of Chemistry and Inano Center University of Aarhus Denmark SAXS Lab
Form and Structure Factors: Modeling and Interactions Jan Skov Pedersen, Department of Chemistry and iNANO Center University of Aarhus Denmark SAXS lab 1 Outline • Model fitting and least-squares methods • Available form factors ex: sphere, ellipsoid, cylinder, spherical subunits… ex: polymer chain • Monte Carlo integration for form factors of complex structures • Monte Carlo simulations for form factors of polymer models • Concentration effects and structure factors Zimm approach Spherical particles Elongated particles (approximations) Polymers 2 Motivation - not to replace shape reconstruction and crystal-structure based modeling – we use the methods extensively - alternative approaches to reduce the number of degrees of freedom in SAS data structural analysis (might make you aware of the limited information content of your data !!!) - provide polymer-theory based modeling of flexible chains - describe and correct for concentration effects 3 Literature Jan Skov Pedersen, Analysis of Small-Angle Scattering Data from Colloids and Polymer Solutions: Modeling and Least-squares Fitting (1997). Adv. Colloid Interface Sci. , 70 , 171-210. Jan Skov Pedersen Monte Carlo Simulation Techniques Applied in the Analysis of Small-Angle Scattering Data from Colloids and Polymer Systems in Neutrons, X-Rays and Light P. Lindner and Th. Zemb (Editors) 2002 Elsevier Science B.V. p. 381 Jan Skov Pedersen Modelling of Small-Angle Scattering Data from Colloids and Polymer Systems in Neutrons, X-Rays and Light P. Lindner and Th. Zemb (Editors) 2002 Elsevier -

The Path Integral Approach to Quantum Mechanics Lecture Notes for Quantum Mechanics IV
The Path Integral approach to Quantum Mechanics Lecture Notes for Quantum Mechanics IV Riccardo Rattazzi May 25, 2009 2 Contents 1 The path integral formalism 5 1.1 Introducingthepathintegrals. 5 1.1.1 Thedoubleslitexperiment . 5 1.1.2 An intuitive approach to the path integral formalism . .. 6 1.1.3 Thepathintegralformulation. 8 1.1.4 From the Schr¨oedinger approach to the path integral . .. 12 1.2 Thepropertiesofthepathintegrals . 14 1.2.1 Pathintegralsandstateevolution . 14 1.2.2 The path integral computation for a free particle . 17 1.3 Pathintegralsasdeterminants . 19 1.3.1 Gaussianintegrals . 19 1.3.2 GaussianPathIntegrals . 20 1.3.3 O(ℏ) corrections to Gaussian approximation . 22 1.3.4 Quadratic lagrangians and the harmonic oscillator . .. 23 1.4 Operatormatrixelements . 27 1.4.1 Thetime-orderedproductofoperators . 27 2 Functional and Euclidean methods 31 2.1 Functionalmethod .......................... 31 2.2 EuclideanPathIntegral . 32 2.2.1 Statisticalmechanics. 34 2.3 Perturbationtheory . .. .. .. .. .. .. .. .. .. .. 35 2.3.1 Euclidean n-pointcorrelators . 35 2.3.2 Thermal n-pointcorrelators. 36 2.3.3 Euclidean correlators by functional derivatives . ... 38 0 0 2.3.4 Computing KE[J] and Z [J] ................ 39 2.3.5 Free n-pointcorrelators . 41 2.3.6 The anharmonic oscillator and Feynman diagrams . 43 3 The semiclassical approximation 49 3.1 Thesemiclassicalpropagator . 50 3.1.1 VanVleck-Pauli-Morette formula . 54 3.1.2 MathematicalAppendix1 . 56 3.1.3 MathematicalAppendix2 . 56 3.2 Thefixedenergypropagator. 57 3.2.1 General properties of the fixed energy propagator . 57 3.2.2 Semiclassical computation of K(E)............. 61 3.2.3 Two applications: reflection and tunneling through a barrier 64 3 4 CONTENTS 3.2.4 On the phase of the prefactor of K(xf ,tf ; xi,ti) .... -

Direct Phase Determination in Protein Electron Crystallography
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 1791–1794, March 1997 Biophysics Direct phase determination in protein electron crystallography: The pseudo-atom approximation (electron diffractionycrystal structure analysisydirect methodsymembrane proteins) DOUGLAS L. DORSET Electron Diffraction Department, Hauptman–Woodward Medical Research Institute, Inc., 73 High Street, Buffalo, NY 14203-1196 Communicated by Herbert A. Hauptman, Hauptman–Woodward Medical Research Institute, Buffalo, NY, December 12, 1996 (received for review October 28, 1996) ABSTRACT The crystal structure of halorhodopsin is Another approach to such phasing problems, especially in determined directly in its centrosymmetric projection using cases where the structures have appropriate distributions of 6.0-Å-resolution electron diffraction intensities, without in- mass, would be to adopt a pseudo-atom approach. The concept cluding any previous phase information from the Fourier of using globular sub-units as quasi-atoms was discussed by transform of electron micrographs. The potential distribution David Harker in 1953, when he showed that an appropriate in the projection is assumed a priori to be an assembly of globular scattering factor could be used to normalize the globular densities. By an appropriate dimensional re-scaling, low-resolution diffraction intensities with higher accuracy than these ‘‘globs’’ are then assumed to be pseudo-atoms for the actual atomic scattering factors employed for small mol- normalization of the observed structure factors. After this ecule structures