On the Role of Cd24 in the Pathogenicity of Myelin Antigen Specific T Cells
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Impact of Action Research Teams on Administrators' Implementation
IMPACT OF ACTION RESEARCH TEAMS ON ADMINISTRATORS’ IMPLEMENTATION AND TEACHERS’ PERCEPTION OF THE TEACHER KEYS EFFECTIVENESS SYSTEM by KEVIN PAUL CARPENTER (Under the Direction of Sheneka Williams) ABSTRACT This action research (AR) case study addresses the effectiveness of the feedback administrators provide to teachers under the Teacher Keys Effectiveness System (TKES). AR teams explored techniques to improve feedback to help teachers improve their instructional practices within teacher and leader efficacy and professional school structures lenses. To evaluate the AR process, the following questions drove the study: 1. How effective is the action research process in empowering the team’s ability to find a solution to an evaluation system problem? 2. How can teachers influence the feedback process, and in what ways does feedback inform teachers’ practices? 3. How do administrators increase their capacity to give actionable feedback? Under the guidance of action research, two teams examined administrator feedback in the TKES platform and the professional learning resources leaders used to improve their evaluation practices. Findings included the following: teachers desire to see consistency in feedback that includes commendations and recommendations, administrators should change an evaluation score if the teacher provides evidence, teachers change their instruction if the feedback is purposeful, relevant, and convincing, and action research teams should include teachers from different grade levels and content areas to be effective as -

OBJ (Application/Pdf)
www.morehouse.edu/themaroontiger Vol. LXXXII, Issue 24 THE MARRON TIGER Morehouse College . Atlanta, GA April 23 - 29, 2008 The Organ of Student Expression Since 1925 Politics as Usual 'Underdog' Mance Wins SGA Presidency; Jonathan Collins Contributing Writer Elected with 54% of the Vote [email protected] pparently Barack Obama started a rev olution. Almost every political plat Aform in the 2008 Morehouse College polit ical elections was based around "change." The question I asked myself is, "When will the need for change end?" It's difficult to comprehend how multiple candidates can preach how a new change is in order on an annual basis. Either their predecessors have not met adequate standards; these new political headliners have such inno vative ideas or its all politics as usual. Although extensive research has not been done, it is pretty certain that student elections have gone on for years. Many changes have been implemented through the election of student officials, but there is only so much change that can occur over a period of time. The problem with many of these "change" platforms is that they under mine the work of the antecedents. Barack Obama never debilitates the works of Dr. Martin Luther King Jr. in his campaign. So, why does a man have to cripple the accom plishments-or lack of-of the man holding the office before him by emphasizing how much "change" he will implement? I guess one must do whatever it takes to become a class officer these days. The interesting thing about these elec tions is how it allows for so many different words to say the same thing. -

Travis Sidak Thesis 2020. Making up a Drug Epidemic
MAKING UP A DRUG EPIDEMIC: CONSTRUCTING DRUG DISCOURSE DURING THE OPIOID EPIDEMIC IN ONTARIO By: Travis M. Sidak Bachelor of Art: Rhetoric and Writing, University of Winnipeg, 2017 A thesis presented to Ryerson University and York University In partial fulfillment of the requirements for the degree of Masters of Arts in the program of Communication and Culture Toronto, Ontario, Canada, 2020 © Travis M. Sidak, 2020 AUTHOR'S DECLARATION FOR ELECTRONIC SUBMISSION OF A THESIS I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I authorize Ryerson University to lend this thesis to other institutions or individuals for the purpose of scholarly research. I further authorize Ryerson University to reproduce this thesis by photocopying or by other means, in total or in part, at the request of other institutions or individuals for the purpose of scholarly research. I understand that my thesis may be made electronically available to the public. i i Making Up a Drug Epidemic: Constructing Drug Discourse During the Opioid Epidemic in Ontario Master of Arts 2020 Travis M. Sidak Communication and Culture Ryerson University & York University Abstract The current opioid epidemic has resulted in growing rates of overdose across the province with the introduction of fentanyl into illicit drug markets. What barriers are preventing policy makers from enacting emergency measures to save lives and how have those affected by the epidemic been categorically ignored? The following research critically analyzes drug discourse relating to the current opioid epidemic in Ontario and discusses why government responses to the epidemic have been delayed, and why they offer inferior measures to prevent growing mortality and morbidity. -

FALL 2020 When I Was Little, I Got a Black Dog Beanie Baby in a Mcdonald’S Happy Meal
FALL 2020 When I was little, I got a black dog beanie baby in a McDonald’s happy meal. I don’t know why I became so attached to it but I did. I named it Nightingale and pretended that the little stuffed black dog would find himself in a series of strange silly predicaments. Once we moved, Nightingale got packed into an unmarked box that very easily could’ve contained anything from old sewing supplies that we’d never reopen, to the dismantled segments of Dad’s drum set. Our new house had a lot more property to play on, more for a young boy to explore ERGO is the literary and artistic publication of Alfred State. and keep himself occupied, so at this It is funded by the Student Senate and is freely distributed point Nightingale had almost entirely each semester. Students, faculty, and staff of Alfred State are slipped from my mind. I spent most of invited to submit their original works of art, poetry, and prose by my time away from my toys exploring e-mailing their submissions to [email protected]. the wilderness (the woods in front of my house), making houses (stick teepees) and Our thanks to everyone who participated this semester and keep hopping ravines (little ditches dug by the submissions coming! little trickling streams). As I grew into my early teens, it became building fires, Sincerely, The Ergo staff exploring the woods beyond the road, and - Susan Perry camping out. When I turned eighteen, I spent a few months backpacking across Europe with my friends, and later traveled around California. -

Neurocognitive Effects of Gist Reasoning Training in Student
NEUROCOGNITIVE EFFECTS OF GIST REASONING TRAINING IN STUDENT- ATHLETES WITH CONCUSSIONS, ADHD, AND LEARNING DISABILITIES Thomas Nguyen, M.S. Dissertation Prepared for the Degree of DOCTOR OF PHILOSOPHY UNIVERSITY O F NORTH TEXAS August 2017 APPROVED: Jennifer Callahan, Committee Co-Chair Trent A. Petrie, Committee Co-Chair Charles Guarnaccia, Committee Member Vicki L. Campbell, Chair of the Department of Psychology David Holdeman, Dean of the College of Arts and Sciences Victor Prybutok, Dean of the Toulouse Graduate School Nguyen, Thomas. Neurocognitive Effects of Gist Reasoning Training in Student-Athletes with Concussions, ADHD, and Learning Disabilities. Doctor of Philosophy (Counseling Psychology), August 2017, 94 pp., 5 tables, references, 154 titles. Concussions, attention-deficit disorder (ADHD), and learning disabilities can adversely impact learning and academic achievement, particularly with respect to attention, memory, and executive functioning; fortunately, cognitive training can be beneficial and remediating these weaknesses. One such program, strategic memory advanced reasoning training (SMART), utilizes a top-down approach to train individuals in executive, higher-ordered thinking strategies including strategic attention, integration, and innovation to facilitate information synthesis and enhance cognitive efficiency. Thus, the purpose of the study is to examine whether SMART improved performances on various neuropsychological measures tapping into attention, processing speed, memory, and executive functioning for college student-athletes with neurological conditions (e.g., concussions, ADHD, LD). Copyright 2017 by Thomas Nguyen ii ACKNOWLEDGEMENTS I would like to express my greatest of appreciation to my mother, father, and sister for consistently being by my side, through the good times and the bad. Without their love and support, I would not be where I am today and for that, I owe them my life. -

Manhood 2.0: a Curriculum Promoting a Gender-Equitable Future of Manhood
A Manhood 2.0: A Curriculum Promoting a Gender-Equitable Future of Manhood Manhood 2.0 is coordinated by Promundo, in partnership with the University of Pittsburgh School of Medicine. Promundo-US 1367 Connecticut Avenue, NW Suite 310 Washington, DC 20036 www.promundoglobal.org University of Pittsburgh School of Medicine 3550 Terrace St. Pittsburgh, PA 15213 www.medschool.pitt.edu Acknowledgements Technical experts from Promundo-US and the University of Pittsburgh Medical Center co-authored the development and adaptation of Manhood 2.0 to the United States. Technical experts from Promundo-US included Dr. Gary Barker, Aapta Garg, Jane Kato-Wallace, Andrew Levack, Ché Nembhard, Magaly Marques, Giovanna Lauro, Natko Geres, Kate Doyle, and Joseph Vess. Technical experts from the University of Pittsburgh included Dr. Elizabeth Miller, Zabi Mulwa, Michael Massof, Paul Mulbah, Nayck Feliz, and Irving Torres. Additional technical contributions came from Amy Fasula, Anna Brittain, Lee Warner, Emilia Koumans, Katherine Kortsmit, and Kathryn Ports from the Centers for Disease Control and Prevention. The co-authors would like to thank Danny Mervil, Tyvon Hewitt, Claudia Blanco, Kelsey Norton, Tina Sojat, and George Garcia from the Latin American Youth Center and Jenita Parkekh, Jennifer Manlove, and Makedah Johnson from ChildTrends for their invaluable insights into the adaptation of Manhood 2.0. Special thanks also go Alexa Hassink, Annaick Miller, and Nina Ford from Promundo for coordinating the production of this curriculum. Copyediting was done by Jill Merriman, and layout was done by Daniel Feary. This project was made possible by RFA-DP-15-007, a partnership between the Teen Pregnancy Prevention Program at the Office of Adolescent Health, U.S. -

Karaoke Catalog Updated On: 15/10/2018 Sing Online on in English Karaoke Songs
Karaoke catalog Updated on: 15/10/2018 Sing online on www.karafun.com In English Karaoke Songs 'Til Tuesday What Can I Say After I Say I'm Sorry Someday You'll Want Me To Want You Voices Carry When You're Smiling (The Whole World Smiles With That Old Black Magic (Woman Voice) (H?D) Planet Earth 1930s Standards That Old Black Magic (Man Voice) Blackout Heartaches I Know Why (And So Do You) DUET Other Side Cheek to Cheek Aren't You Glad You're You 10 Years My Romance (I've Got A Gal In) Kalamazoo Through The Iris It's Time To Say Aloha No Love No Nothin' 10,000 Maniacs We Gather Together Personality Because The Night Kumbaya Sunday, Monday Or Always 10CC The Last Time I Saw Paris This Heart Of Mine Dreadlock Holiday All The Things You Are Mister Meadowlark I'm Not In Love Smoke Gets In Your Eyes 1950s Standards The Things We Do For Love Begin The Beguine Get Me To The Church On Time Rubber Bullets I Love A Parade Fly Me To The Moon Life Is A Minestrone I Love A Parade (short version) It's Beginning To Look A Lot Like Christmas 112 I'm Gonna Sit Right Down And Write Myself A Letter Crawdad Song Cupid Body And Soul Christmas In Killarney Peaches And Cream Man On The Flying Trapeze That's Amore 12 Gauge Pennies From Heaven My Own True Love (Tara's Theme) Dunkie Butt When My Ship Comes In Organ Grinder's Swing 12 Stones Yes Sir, That's My Baby Lullaby Of Birdland Far Away About A Quarter To Nine Rags To Riches Crash Did You Ever See A Dream Walking? Something's Gotta Give 1800s Standards I Thought About You I Saw Mommy Kissing Santa Claus (Man -

The Rational Male Free
FREE THE RATIONAL MALE PDF Rollo Tomassi | 302 pages | 01 Oct 2013 | Createspace Independent Publishing Platform | 9781492777861 | English | United States The Rational Male - Positive Masculinity by Rollo Tomassi Some of it might end you hating women instead of realizing how they work. However, in this rational male summary, I will only put my thoughts of it at the The Rational Male of the post and highlighted in between. Disclaimer : I still recommend reading the whole book. You might get the message wrong. See it as a glimpse into it or to cement your buying decision. It basically revolves around desire and power dynamics which have been shifted to the feminine in our society. Being a man is not inherently bad. The Rational Male book goes over the reasons why that is. Why women go for jerks, even though they always say differently. It teaches you the necessary information about both sexes and how to navigate the sexual marketplace. Plus, mistakes you have been making in your approach and living with women in general. This term describes a man being obsessed with one woman which a lot of guys seem to have these days. In short: There are almost 4 billion women out there, why do you think only this one will do? And a lot of guys fall into that trap with just a random woman they happen to get into, just because she probably took a few facials, he feels like she is the one. The secret to relationships lies in one simple power dynamic which has been touched by other Authors multiple times: The person with the least interest in the relationship has the power. -

Sermon 22 Aug 2021
JESUS, BROTHER OF JAMES FIRST PRESBYTERIAN CHURCH OF GREENWOOD 22 AUGUST 2021 REV. GORDON TUBBS There are quite a few blockbuster movies in the theaters right now that you may be aware of. For a big movie buff like me, it’s nice to have a bunch of options, but that just creates another problem: which one am I going to prioritize seeing over the other. You may be familiar with this problem, it’s known as analysis paralysis. And one proven strategy to solve this problem is to simplify and narrow down your choices to their essential features. In fact there are three ways we normally do this when it comes to movies. The first way is to read the synopsis, the second way is to see what genre the film is, and the third way is to check the critic ratings. But if you’re on Twitter or social media, you’ve no doubt seen posts of people providing a short synopsis that’s so bad it’s actually good. Take these for example: Lord of the Rings Trilogy Group spends 9 hours returning a ring to a volcano. Titanic Rich girl lets a poor boy freeze to death. Star Wars Saga One family’s dysfunction ruins the galaxy. Beauty and the Beast Good girl meets a bad boy and thinks she can fix him up. Jaws Big shark bites a small boat. Then they get a bigger boat. Casablanca Man is torn between a girl and the war-effort. My Fair Lady Girl learns to speak in an accent to make it in a new town. -
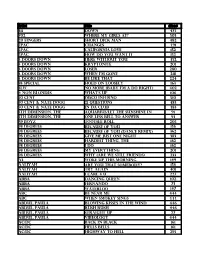
Music List (PDF)
Artist Title disc# 311 Down 475 702 Where My Girls At? 503 20 fingers short dick man 482 2pac changes 491 2Pac California Love 152 2Pac How Do You Want It 152 3 Doors Down Here Without You 452 3 Doors Down Kryptonite 201 3 doors down loser 280 3 Doors Down when I'm gone 218 3 Doors Down Be Like That 224 38 Special Hold On Loosely 163 3lw No more (baby I'm a do right) 402 4 Non Blondes What's Up 106 50 Cent Disco Inferno 510 50 cent & nate dogg 21 questions 483 50 cent & nate dogg in da club 483 5th Dimension, The aquarius/let the sunshine in 91 5th Dimension, The One Less Bell To Answer 93 69 Boyz Tootsee Roll 205 98 Degrees Because Of You 156 98 Degrees Because Of You (Dance Remix) 362 98 degrees Give me just one Night 383 98 Degrees Hardest Thing, the 162 98 Degrees I Do 162 98 Degrees My Everything 201 98 Degrees Why (Are We Still Friends) 233 A3 Woke Up This Morning 149 Aaliyah Are You That Somebody? 156 aaliyah Try again 401 Aaliyah I Care 4 U 227 Abba Dancing Queen 102 ABBA FERNANDO 73 ABBA Waterloo 147 ABC Be Near Me 444 ABC When Smokey Sings 444 ABDUL, PAULA Blowing Kisses In The Wind 446 ABDUL, PAULA Rush Rush 446 ABDUL, PAULA STRAIGHT UP 27 ABDUL, PAULA Vibeology 444 AC/DC Back In Black 161 AC/DC Hells Bells 161 AC/DC Highway To Hell 295 AC/DC Stiff Upper Lip 295 AC/DC Whole Lotta Rosie 295 AC/dc You shook me all night long 108 Ace Of Base Beautiful Life 8 Ace of Base Don't Turn Around 154 Ace Of Base Sign, The 206 AD LIBS BOY FROM NEW YORK CITY, THE 49 adam ant goody two shoes 108 ADAMS, BRYAN (EVERYTHING I DO) I DO IT FOR YOU -

DJ Song List
978-256-1899 DJ Example List BY ARTIST Artist Title Disc Poloma Polka 203 A 03 Pennsylvania Polka 203 A 06 My Melody Of Love Polka 203 A 10 Mexican Hat Dance 201 B 07 La Raspa 201 B 01 Just Because 203 A 09 It’s A Small World Polka 203 A 08 Hava Nagila 202 A 04 Happy Birthday 202 A 01 Domino Polka 203 A 02 Clarinet Polka 203 A 07 Cherry Pink & Apple Blossom 201 B 02 Champaigne Polka 203 A 02 Bunny Hop 201 B 08 Beer Barrel Polka 203 A 05 Baruska 203 A 04 1 Giant Leap feat My Culture POct02-12 1. PlusWilliams 1 CherryMy Culture Bomb T552-11 1000 Clowns (Not The) Greatest Rapper T470-13 10,000 Maniacs More Than This T382-06 10,000 Maniacs More Than This RH 4312 10,000 Maniacs Rainy Day (Edit) T403-11 10,000 Maniacs Trouble Me 89 A 06 112 Come See Me T362-16 112 Cupid (Radio Mix) T376-08 112 Only You T330-15 112 Your Letter (Radio Mix) T491-08 112 It’s Over Now (Radio Edit) T563-09 112 Peaches & Cream PJuly01-17 112 (f/ Lil' Z) Anywhere (Radio Mix) T465-12 112 f/Mase Love Me (Radio Mix) T454-11 12 Volt Sex Hook It Up T557-14 15 Distracted T549-19 1910 Fruitgum Compa Simon Says 205 B 03 2 Elvissa Oh La La La (Radio Mix) T392-12 2 Live Crew Banned In The Sea 94 B 04 2 Pac Changes (Radio Edit) T459-10 2 Pac I Wonder If Heaven Got A Ghetto T408-13 2 Pac F/KC & Jo Jo How Do You Want It T334-14 2 Pistols f./T-Pain & Dizm She Got It *Promo Only Clean Edit* PFeb08-10 April 23, 2013 Page 1 2 Skinee J’s Grown Up PApril02-16 2 Unlimited Get Ready For This Dance Mix 4-14 2 Unlimited Tribal Dance Dance Mix USA 15 2 Unlimited Twilight Zone Dance Mix USA -

Canciones De Karaoke Actualizado El: 06/07/2015 Canta En Línea En Todo El Catálogo
Canciones de karaoke Actualizado el: 06/07/2015 Canta en línea en www.karafun.es Todo el catálogo Los 50 mejores Bailando - Enrique Iglesias Libre soy - Frozen Mi Nuevo Vicio - Paulina Rubio Vivir mi vida - Marc Anthony Burbujas De Amor - Juan Luis Guerra Rolling In The Deep - Adele El Taxi - Pitbull Darte un beso - Prince Royce Limbo - Daddy Yankee El perdón - Nicky Jam Sabor A Mi - Luis Miguel All Of Me - John Legend Propuesta Indecente - Romeo Santos Me va, me va - Julio Iglesias New York, New York - Frank Sinatra Un beso y una flor - Nino Bravo Vivir sin aire - Maná Thinking Out Loud - Ed Sheeran Corazón partío - Alejandro Sanz Y como es el - José Luis Perales A fuego lento - Rosana La camisa negra - Juanes Color esperanza - Diego Torres Cuando Me Enamoro - Enrique Iglesias Colgando en tus manos (duo) - Carlos Baute Maria (Un, dos, tres) - Ricky Martin No Me Ames - Marc Anthony Mi gran noche - Rafael Martos Sánchez My Way - Frank Sinatra A dios le pido - Juanes Loco - Enrique Iglesias Humanos a marte - Chayanne Summer Nights - Grease Happy - Pharrell Williams Hijo de la luna - Mecano El porompompero - Manolo Escobar Mi verdad - Maná La Bomba - King Africa Obsesión - Aventura Danza kuduro - Don Omar Por eso te canto - Melendi It's Raining Men - Geri Halliwell Uptown Funk - Bruno Mars Macarena (Spanish Version) - Los Del Rio Besame Mucho - Andrea Bocelli La Vida Es Un Carnaval - Celia Cruz Ojalá que llueva café - Juan Luis Guerra Yesterday - The Beatles Mi Niña Bonita - Chino & Nacho Valió la pena (Salsa version) - Marc Anthony EXPLíCITO