Cytochrome P450 Oxidoreductase Deficiency in Three Patients Initially
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Altered Expression and Function of Mitochondrial Я-Oxidation Enzymes
0031-3998/01/5001-0083 PEDIATRIC RESEARCH Vol. 50, No. 1, 2001 Copyright © 2001 International Pediatric Research Foundation, Inc. Printed in U.S.A. Altered Expression and Function of Mitochondrial -Oxidation Enzymes in Juvenile Intrauterine-Growth-Retarded Rat Skeletal Muscle ROBERT H. LANE, DAVID E. KELLEY, VLADIMIR H. RITOV, ANNA E. TSIRKA, AND ELISA M. GRUETZMACHER Department of Pediatrics, UCLA School of Medicine, Mattel Children’s Hospital at UCLA, Los Angeles, California 90095, U.S.A. [R.H.L.]; and Departments of Internal Medicine [D.E.K., V.H.R.] and Pediatrics [R.H.L., A.E.T., E.M.G.], University of Pittsburgh School of Medicine, Magee-Womens Research Institute, Pittsburgh, Pennsylvania 15213, U.S.A. ABSTRACT Uteroplacental insufficiency and subsequent intrauterine creased in IUGR skeletal muscle mitochondria, and isocitrate growth retardation (IUGR) affects postnatal metabolism. In ju- dehydrogenase activity was unchanged. Interestingly, skeletal venile rats, IUGR alters skeletal muscle mitochondrial gene muscle triglycerides were significantly increased in IUGR skel- expression and reduces mitochondrial NADϩ/NADH ratios, both etal muscle. We conclude that uteroplacental insufficiency alters of which affect -oxidation flux. We therefore hypothesized that IUGR skeletal muscle mitochondrial lipid metabolism, and we gene expression and function of mitochondrial -oxidation en- speculate that the changes observed in this study play a role in zymes would be altered in juvenile IUGR skeletal muscle. To test the long-term morbidity associated with IUGR. (Pediatr Res 50: this hypothesis, mRNA levels of five key mitochondrial enzymes 83–90, 2001) (carnitine palmitoyltransferase I, trifunctional protein of -oxi- dation, uncoupling protein-3, isocitrate dehydrogenase, and mi- Abbreviations tochondrial malate dehydrogenase) and intramuscular triglycer- CPTI, carnitine palmitoyltransferase I ides were quantified in 21-d-old (preweaning) IUGR and control IUGR, intrauterine growth retardation rat skeletal muscle. -

Isocitrate Dehydrogenase 1 (NADP+) (I5036)
Isocitrate Dehydrogenase 1 (NADP+), human recombinant, expressed in Escherichia coli Catalog Number I5036 Storage Temperature –20 °C CAS RN 9028-48-2 IDH1 and IDH2 have frequent genetic alterations in EC 1.1.1.42 acute myeloid leukemia4 and better understanding of Systematic name: Isocitrate:NADP+ oxidoreductase these mutations may lead to an improvement of (decarboxylating) individual cancer risk assessment.6 In addition other studies have shown loss of IDH1 in bladder cancer Synonyms: IDH1, cytosolic NADP(+)-dependent patients during tumor development suggesting this may isocitrate dehydrogenase, isocitrate:NADP+ be involved in tumor progression and metastasis.7 oxidoreductase (decarboxylating), Isocitric Dehydrogenase, ICD1, PICD, IDPC, ICDC, This product is lyophilized from a solution containing oxalosuccinate decarboxylase Tris-HCl, pH 8.0, with trehalose, ammonium sulfate, and DTT. Product Description Isocitrate dehydrogenase (NADP+) [EC 1.1.1.42] is a Purity: ³90% (SDS-PAGE) Krebs cycle enzyme, which converts isocitrate to a-ketoglutarate. The flow of isocitrate through the Specific activity: ³80 units/mg protein glyoxylate bypass is regulated by phosphorylation of isocitrate dehydrogenase, which competes for a Unit definition: 1 unit corresponds to the amount of 1 common substrate (isocitrate) with isocitrate lyase. enzyme, which converts 1 mmole of DL-isocitrate to The activity of the enzyme is dependent on the a-ketoglutarate per minute at pH 7.4 and 37 °C (NADP formation of a magnesium or manganese-isocitrate as cofactor). The activity is measured by observing the 2 complex. reduction of NADP to NADPH at 340 nm in the 7 presence of 4 mM DL-isocitrate and 2 mM MnSO4. -

Pro-Aging Effects of Xanthine Oxidoreductase Products
antioxidants Review Pro-Aging Effects of Xanthine Oxidoreductase Products , , Maria Giulia Battelli y , Massimo Bortolotti y , Andrea Bolognesi * z and Letizia Polito * z Department of Experimental, Diagnostic and Specialty Medicine-DIMES, Alma Mater Studiorum, University of Bologna, Via San Giacomo 14, 40126 Bologna, Italy; [email protected] (M.G.B.); [email protected] (M.B.) * Correspondence: [email protected] (A.B.); [email protected] (L.P.); Tel.: +39-051-20-9-4707 (A.B.); +39-051-20-9-4729 (L.P.) These authors contributed equally. y Co-last authors. z Received: 22 July 2020; Accepted: 4 September 2020; Published: 8 September 2020 Abstract: The senescence process is the result of a series of factors that start from the genetic constitution interacting with epigenetic modifications induced by endogenous and environmental causes and that lead to a progressive deterioration at the cellular and functional levels. One of the main causes of aging is oxidative stress deriving from the imbalance between the production of reactive oxygen (ROS) and nitrogen (RNS) species and their scavenging through antioxidants. Xanthine oxidoreductase (XOR) activities produce uric acid, as well as reactive oxygen and nitrogen species, which all may be relevant to such equilibrium. This review analyzes XOR activity through in vitro experiments, animal studies and clinical reports, which highlight the pro-aging effects of XOR products. However, XOR activity contributes to a regular level of ROS and RNS, which appears essential for the proper functioning of many physiological pathways. This discourages the use of therapies with XOR inhibitors, unless symptomatic hyperuricemia is present. -

RESEARCH COMMUNICATION HADHA Is a Potential Predictor Of
HADHA is a Potential Predictor of the Response to Platinum-based Chemotherapy RESEARCH COMMUNICATION HADHA is a Potential Predictor of Response to Platinum-based Chemotherapy for Lung Cancer Taihei Kageyama1, Ryo Nagashio1, 2, Shinichiro Ryuge 3, Toshihide Matsumoto1,5, Akira Iyoda4, Yukitoshi Satoh4, Noriyuki Masuda3, Shi-Xu Jiang5, Makoto Saegusa5, Yuichi Sato1, 2* Abstract To identify a cisplatin resistance predictor to reduce or prevent unnecessary side effects, we firstly established four cisplatin-resistant sub-lines and compared their protein profiles with cisplatin-sensitive parent lung cancer cell lines using two-dimensional gel electrophoresis. Between the cisplatin-resistant and -sensitive cells, a total of 359 protein spots were differently expressed (>1.5 fold), and 217 proteins (83.0%) were identified. We focused on a mitochondrial protein, hydroxyl-coenzyme A dehydrogenase/3-ketoacyl-coenzyme A thiolase/enoyl-coenzyme A hydratase alpha subunit (HADHA), which was increased in all cisplatin-resistant cells. Furthermore, pre- treated biopsy specimens taken from patients who showed resistance to platinum-based treatment showed a significantly higher positive rate for HADHA in all cases (p=0.00367), including non-small cell lung carcinomas (p=0.002), small-cell lung carcinomas (p=0.038), and adenocarcinomas (p=0.008). These results suggest that the expression of HADHA may be a useful marker to predict resistance to platinum-based chemotherapy in patients with lung cancer. Keywords: Cisplatin - HADHA - lung cancer - two-dimensional gel electrophoresis Asian Pacific J Cancer Prev, 12, 3457-3463 Introduction cisplatin resistance rose due to a decrease of blood flow in the tumor and increased DNA repair (Stewart, 2007), Lung cancer is the leading cause of cancer-related the mechanisms underlying cisplatin resistance have not death in the world, and the five-year overall survival rate yet been clarified, and an effective cisplatin resistance is still below 16% (Jemal et al., 2009). -

139 Normal Bone Density in Male
SEPTEMBER-OCTOBER REV. HOSP. CLÍN. FAC. MED. S. PAULO 56(5):139-142, 2001 NORMAL BONE DENSITY IN MALE PSEUDOHERMAPHRODITISM DUE TO 5α- REDUCTASE 2 DEFICIENCY Elaine Maria Frade Costa, Ivo Jorge Prado Arnhold, Marlene Inacio and Berenice Bilharinho Mendonca RHCFAP/3050 COSTA EMF et al. - Normal bone density in male pseudohermaphroditism due to 5α-reductase 2 deficiency. Rev. Hosp. Clín. Fac. Med. S. Paulo 56(5):139-142, 2001. Bone is an androgen-dependent tissue, but it is not clear whether the androgen action in bone depends on testosterone or on dihydrotestosterone. Patients with 5α-reductase 2 deficiency present normal levels of testosterone and low levels of dihydrotestosterone, providing an in vivo human model for the analysis of the effect of testosterone on bone. Objective: To analyze bone mineral density in 4 adult patients with male pseudohermaphroditism due to 5α-reductase 2 deficiency. Results: Three patients presented normal bone mineral density of the lumbar column (L1-L4) and femur neck, and the other patient presented a slight osteopenia in the lumbar column. Conclusion: Patients with dihydrotestosterone deficiency present normal bone mineral density, suggesting that dihydrotestosterone is not the main androgen acting in bone. DESCRIPTOR: Bone mineral density. Male pseudohermaphroditism. 5α-reductase type 2 deficiency. It has been well documented in the fied androgenic receptors in these cells, fects require aromatization into estro- literature that gonadal steroids regulate thus demonstrating that both androgens gens with subsequent activation of the normal bone metabolism and that in- and estrogens act by a direct mecha- estrogenic receptor. Although it has adequate estrogen concentrations in fe- nism through their respective receptors. -

400 We Have Studied Six Infants and Young Children with Hyper
400 ABSTRACTS We have studied six infants and young children with hyper- ml plasma sample has been evaluated for the rapid (4—6 hr) diag- thyroidism whose clinical course differs from the few reports of nosis of CAH. Pet ether, benzene and methylene chloride extracts others. Neonatal and early childhood hyperthyroidism are usually of plasma are quantitated by competitive protein binding using thought of as separate, rare, and transient disorders seldom re- 17-hydroxyprogesterone (17-OHP), 11-deoxycortisol (cmpd S), quiring long term treatment. 1) Our cases have not been tran- and cortisol standards, respectively, for comparison. The observed sient: 2) they have occurred in families with a high incidence of plasma steroid concentrations are expressed as a ratio of adult Graves disease. "17-OHP" + "cmpd S" to "cortisol" since comparison of ratios, Four were born with Graves disease. Three continue to be rather than absolute values, has been found to differentiate nor- hyperthyroid at ages 1, 5, and 6 years. Two developed Graves mals from patients more clearly. disease at ages 3 and 8 years and continue on anti-thyroid medi- Plasma samples have been obtained from six normal children cation. Graves disease occurred in five of the six mothers and aged 4 days-7 yrs following administration of ACTH, from six was apparent during gestation in four. The sixth mother, mother adults with 11-hydroxylation impaired by the administration of of a neonatal case, has never had overt Graves disease, but female metyrapone, and from three children aged 11 mos, 6 yrs and 8 members of four generations have had Graves disease. -

Endocrinology Test List Endocrinology Test List
For Endocrinologists Endocrinology Test List Endocrinology Test List Extensive Capabilities Managing patients with endocrine disorders is complex. Having access to the right test for the right patient is key. With a legacy of expertise in endocrine laboratory diagnostics, Quest Diagnostics offers an extensive menu of laboratory tests across the spectrum of endocrine disorders. This test list highlights the extensive menu of laboratory diagnostic tests we offer, including highly specialized tests and those performed using highly specific and sensitive mass spectrometry detection. It is conveniently organized by glandular function or common endocrine disorder, making it easy for you to identify the tests you need to care for the patients you treat. Comprehensive Care Quest Diagnostics Nichols Institute has been pioneering state-of-the-art endocrine testing for over four decades. Our commitment to innovative diagnostics and our dedication to quality and service means we deliver solutions that enable you to make informed clinical decisions for comprehensive patient management. We strive to remain at the forefront of innovation in endocrine testing so you can deliver the highest level of patient care. Abbreviations and Footnotes NDM, neonatal diabetes mellitus; MODY, maturity-onset diabetes of the young; CH, congenital hyperinsulinism; MSUD, maple syrup urine disease; IHH, idiopathic hypogonadotropic hypogonadism; BBS, Bardet-Biedl syndrome; OI, osteogenesis imperfecta; PKD, polycystic kidney disease; OPPG, osteoporosis-pseudoglioma syndrome; CPHD, combined pituitary hormone deficiency; GHD, growth hormone deficiency. The tests highlighted in green are performed using highly specific and sensitive mass spectrometry detection. Panels that include a test(s) performed using mass spectrometry are highlighted in yellow. For tests highlighted in blue, refer to the Athena Diagnostics website (athenadiagnostics.com/content/test-catalog) for test information. -

CYP19A1 Gene Cytochrome P450 Family 19 Subfamily a Member 1
CYP19A1 gene cytochrome P450 family 19 subfamily A member 1 Normal Function The CYP19A1 gene provides instructions for making an enzyme called aromatase. This enzyme converts a class of hormones called androgens, which are involved in male sexual development, to different forms of the female sex hormone estrogen. In cells, aromatase is found in a structure called the endoplasmic reticulum, which is involved in protein production, processing, and transport. The activity (expression) of aromatase varies among different cell types depending on the cells' need for estrogen. In females, aromatase is most active in the ovaries, where it guides sexual development. In males, aromatase is most active in fat (adipose) tissue. In both males and females, estrogen plays a role in regulating bone growth and blood sugar levels. During fetal development, aromatase converts androgens to estrogens in the placenta, which is the link between the mother's blood supply and the fetus. This conversion in the placenta prevents androgens from directing sexual development in female fetuses. After birth, the conversion of androgens to estrogens takes place in multiple tissues. Health Conditions Related to Genetic Changes Aromatase deficiency More than 20 mutations in the CYP19A1 gene have been found to cause aromatase deficiency. This condition is characterized by reduced levels of estrogen and increased levels of androgens. These abnormal hormone levels lead to impaired sexual development in affected females and unusual bone growth, insulin resistance, and other signs and symptoms in both males and females with the condition. CYP19A1 gene mutations that cause aromatase deficiency decrease or eliminate aromatase activity. A lack of aromatase function results in an inability to convert androgens to estrogens before birth and throughout life. -

Four Clinical Variants of Congenital Adrenal Hyperplasia
Arch Dis Child: first published as 10.1136/adc.39.203.66 on 1 February 1964. Downloaded from Arch. Dis. Childh., 1964, 39, 66. FOUR CLINICAL VARIANTS OF CONGENITAL ADRENAL HYPERPLASIA BY W. HAMILTON and M. G. BRUSH From the University Department of Child Health and the Royal Hospitalfor Sick Children, Glasgow, and the University Department of Steroid Biochemistry and the Royal Infirmary, Glasgow (RECEIVED FOR PUBLICATION AUGUST 26, 1963) Three clinical types of congenital adrenogenital Case Reports virilism due to adrenal hyperplasia have now been Case 1. This child, born January 27, 1954, was of well defined. These are simple virilization, viriliza- ambiguous sex having a curved phallus with an opening tion with excessive sodium loss and danger to life at the tip. Both this and another opening on the peri- and virilization combined with hypertension. Clini- neum admitted a probe to a depth of 1 cm. The scrotum cal subvariants have also been described in asso- was bifid and the testes were not palpated. ciation with hypoglycaemia (White and Sutton, When 5 weeks of age, a skin biopsy and buccal smear examined for sex chromatin indicated that the child was 1951; Wilkins, Crigler, Silverman, Gardner and female. The urinary 17-oxosteroids were reported as Migeon, 1952), with periodic fever (Gonzales and 0 4 mg. per day. When 2 years of age the perineum Gardner, 1956; Gardner and Migeon, 1959) and with was opened up in the midline and the urethral and the late onset of sodium loss (Cara and Gardner, vaginal orifices were found to open on to a small vestibule. -

Glyoxysomal Malate Dehydrogenase from Watermelon Is Synthesized
Proc. Nati. Acad. Sci. USA Vol. 87, pp. 5773-5777, August 1990 Botany Glyoxysomal malate dehydrogenase from watermelon is synthesized with an amino-terminal transit peptide (isoenzymes/organelle/Citrulus vulgaris/polymerase chain reaction) CHRISTINE GIETL* Institute of Botany, Technical University of Munich, Arcisstrasse 16, D-8000 Munich 2, Federal Republic of Germany; and Department of Physiology, Carlsberg Laboratory, Gamle Carlsberg Vej 10, DK-2500 Copenhagen Valby, Denmark Communicated by Diter von Wettstein, May 11, 1990 (receivedfor review March 5, 1990) ABSTRACT The isolation and sequence of a cDNA clone and peroxisomes are seen between mitochondria and chlo- encoding the complete glyoxysomal malate dehydrogenase roplasts (2). As in plants, mammalian peroxisomes contain [gMDH; (S)-malate:NAD+ oxidoreductase, EC 1.1.1.37] of enzymes involved in the production and degradation ofH202; watermelon cotyledons are presented. Partial cDNA clones in trypanosomes, glycolysis is sequestered into microbodies were synthesized in a three part strategy, taking advantage of called glycosomes (3). All microbodies studied contain en- the polymerase chain reaction technology with oligonucleotides zymes for (3-oxidation and a specific spectrum of other based on directly determined amino acid sequences. Subse- enzymes. It is further characteristic that microbody enzyme quently, the complete done for gMDH was synthesized with a activities are also present in other cell compartments. Gly- sense primer corresponding to the nucleotide sequence of the oxysomes as well as mitochondria contain MDH, citrate N-terminal end of pre-gMDIH and an antisense primer corre- synthase, and enzymes for 83-oxidation (4). The organelle- sponding to the adenylylation site found in the mRNA. -

Cholic Acid for Treating Inborn Errors of Primary Bile Acid Synthesis NHS England Unique Reference Number URN1623 / NICE ID004
NATIONAL INSTITUTE FOR HEALTH AND CARE EXCELLENCE Clinical evidence review of cholic acid for treating inborn errors of primary bile acid synthesis NHS England unique reference number URN1623 / NICE ID004 Prepared by: NICE on behalf of NHS England Specialised Commissioning About this clinical evidence review Clinical evidence reviews are a summary of the best available evidence for a single technology within a licensed indication, for commissioning by NHS England. The clinical evidence review supports NHS England in producing clinical policies but is not NICE guidance or advice. Summary This evidence review considers cholic acid (Laboratoires CTRS [Orphacol] and Retrophin Europe Ltd [Kolbam]) for treating inborn errors of primary bile acid NICE clinical evidence review of cholic acid for treating inborn errors of primary bile acid synthesis Page 1 of 72 NHS URN1623 NICE ID004 synthesis caused by the following enzyme deficiencies in people aged 1 month and over: 3-beta-hydroxy-delta5-C27-steroid oxidoreductase (3beta-HSD) delta4-3-oxosteroid-5-beta reductase (5beta-reductase) 2- (or alpha-) methylacyl-CoA racemase (AMACR) sterol 27-hydroxylase (presenting as cerebrotendinous xanthomatosis [CTX]) cholesterol 7alpha-hydroxylase (CYP7A1). Inborn errors of primary bile acid synthesis are rare genetic conditions in which enzyme deficiencies prevent the liver from converting cholesterol in the body to bile acids (such as cholic acid and chenodeoxycholic acid). This results in the liver producing high concentrations of atypical (or ‘unusual’) bile acids and intermediary metabolites (some of which are toxic to the liver) in an attempt to establish a normal bile acid pool. Accumulation of potentially toxic atypical bile acids and metabolites, and reduced flow of bile acids may cause liver injury. -
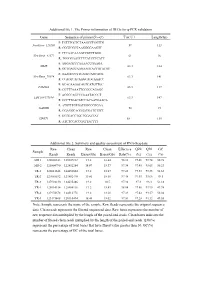
Additional File 1
Additional file 1. The Primer information of DEGs for q-PCR validation Gene Sequence of primer(5'→3') Tm(℃) Length(bp) F: TGTTTGCTCTAAGCCTGGTTG NewGene_126260 59 113 R: CGGTCGCTAAGGGGAAGTT F: CTCAACAAAGCCGTCTGGG NewGene_41572 61 96 R: TGGGGAATCTTCATCCTCATT F: AGGAGCCCAAAACCGAAGA MME 63.3 184 R: GCTGACCAAGAAGTACCGTATGT F: AAAGCCCTTCAGTCAGCACG NewGene_70974 63.3 141 R: CCAGTCACAAGCAGCAAACC F: GCACAAGGCAGTCATGTTGC FAM43A 63.3 117 R: CGTTTAAATTCCGCCAGAGC F: ACGGCAGCCCAAATACCCT LOC108177184 63.3 147 R: GCCTTGACATCCACAATGAACA F: ATGTTTGTGATGGGCGTGAA GAPDH 58 94 R: GGAGGCAGGGATGATGTTCT F: GCTGACCTGCTGGATTAT HPRT1 58 135 R: ATCTCCACCGATTACTTT Additional file 2. Summary and quality assessment of RNA-Seq data Raw Clean Raw Clean Effective Q20 Q30 GC Sample Reads Reads Bases(Gb) Bases(Gb) Rate(%) (%) (%) (%) MR-1 128008342 125607312 19.2 18.84 98.12 97.46 93.74 50.78 MR-2 125804790 122452284 18.87 18.37 97.34 97.45 93.65 50.23 TR-1 128011648 124452604 19.2 18.67 97.22 97.33 93.35 51.65 TR-2 127085532 123882470 19.06 18.58 97.48 97.45 93.66 49.5 TR-3 127994130 124635486 19.2 18.7 97.38 97.3 93.3 51.18 YR-1 128014314 125504116 19.2 18.83 98.04 97.56 93.95 49.98 YR-2 127974678 124512778 19.2 18.68 97.29 97.42 93.57 50.04 YR-3 123173408 120110494 18.48 18.02 97.51 97.28 93.32 49.55 Note: Sample represents the name of the sample. Raw Reads represents the original sequence data. Clean reads represents the filtered sequenced data. Raw bases represents the number of raw sequence data multiplied by the length of the paired-end reads.