Opioid Receptor Binding, Signaling and Antinociceptive Activity
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Alternative Formats If You Require This Document in an Alternative Format, Please Contact: [email protected]
University of Bath PHD The extraction and chemistry of the metabolites of Mimosa tenuiflora and Papaver somniferum Ninan, Aleyamma Award date: 1990 Awarding institution: University of Bath Link to publication Alternative formats If you require this document in an alternative format, please contact: [email protected] General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 23. Sep. 2021 THE EXTRACTION AND CHEMISTRY OF THE METABOLITES OF MIMOSA TENUIFLORA AND PAP AVER SOMNIFERUM. submitted by ALEYAMMA NINAN for the degree of Doctor of Philosophy of the University of Bath 1990 Attention is drawn to the fact that the copyright of this thesis rests with its author. This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with its author and that no quotation from the thesis and no information derived from it may be published without prior consent of the author. -

(12) United States Patent (10) Patent No.: US 9,687,445 B2 Li (45) Date of Patent: Jun
USOO9687445B2 (12) United States Patent (10) Patent No.: US 9,687,445 B2 Li (45) Date of Patent: Jun. 27, 2017 (54) ORAL FILM CONTAINING OPIATE (56) References Cited ENTERC-RELEASE BEADS U.S. PATENT DOCUMENTS (75) Inventor: Michael Hsin Chwen Li, Warren, NJ 7,871,645 B2 1/2011 Hall et al. (US) 2010/0285.130 A1* 11/2010 Sanghvi ........................ 424/484 2011 0033541 A1 2/2011 Myers et al. 2011/0195989 A1* 8, 2011 Rudnic et al. ................ 514,282 (73) Assignee: LTS Lohmann Therapie-Systeme AG, Andernach (DE) FOREIGN PATENT DOCUMENTS CN 101703,777 A 2, 2001 (*) Notice: Subject to any disclaimer, the term of this DE 10 2006 O27 796 A1 12/2007 patent is extended or adjusted under 35 WO WOOO,32255 A1 6, 2000 U.S.C. 154(b) by 338 days. WO WO O1/378O8 A1 5, 2001 WO WO 2007 144080 A2 12/2007 (21) Appl. No.: 13/445,716 (Continued) OTHER PUBLICATIONS (22) Filed: Apr. 12, 2012 Pharmaceutics, edited by Cui Fude, the fifth edition, People's Medical Publishing House, Feb. 29, 2004, pp. 156-157. (65) Prior Publication Data Primary Examiner — Bethany Barham US 2013/0273.162 A1 Oct. 17, 2013 Assistant Examiner — Barbara Frazier (74) Attorney, Agent, or Firm — ProPat, L.L.C. (51) Int. Cl. (57) ABSTRACT A6 IK 9/00 (2006.01) A control release and abuse-resistant opiate drug delivery A6 IK 47/38 (2006.01) oral wafer or edible oral film dosage to treat pain and A6 IK 47/32 (2006.01) substance abuse is provided. -

(Oxycontin®) and Oxymorphone (Opana® ER) Reference Number: ERX.NSST.17 Effective Date: 06/15 Revision Log Last Review Date: 09/16
Clinical Policy: extended-release oxycodone (OxyContin®) and oxymorphone (Opana® ER) Reference Number: ERX.NSST.17 Effective Date: 06/15 Revision Log Last Review Date: 09/16 Clinical policies are intended to be reflective of current scientific research and clinical thinking. This policy is current at the time of approval, may be updated and therefore is subject to change. This Clinical Policy is not intended to dictate to providers how to practice medicine, nor does it constitute a contract or guarantee regarding payment or results. Providers are expected to exercise professional medical judgment in providing the most appropriate care, and are solely responsible for the medical advice and treatment of members. This policy is the property of Envolve Pharmacy Solutions. Unauthorized copying, use, and distribution of this Policy or any information contained herein is strictly prohibited. By accessing this policy, you agree to be bound by the foregoing terms and conditions, in addition to the Site Use Agreement for Health Plans associated with Envolve Pharmacy Solutions. Description The intent of the criteria is to ensure that patients follow selection elements established by Envolve Pharmacy Solutions for the use of extended-release oxycodone (OxyContin®) and oxymorphone (Opana® ER). Policy/Criteria It is the policy of health plans affiliated with Envolve Pharmacy Solutions® that extended-release oxycodone (OxyContin®) and oxymorphone (Opana® ER) are medically necessary for members meeting the following criteria: Initial Approval Criteria (must meet all): A. Failure of an immediate-release (short-acting) narcotic analgesic; B. Failure of fentanyl patch and morphine extended-release tablets/capsules in the past 6 months, unless intolerant or contraindicated; C. -

Veterinary Anesthetic and Analgesic Formulary 3Rd Edition, Version G
Veterinary Anesthetic and Analgesic Formulary 3rd Edition, Version G I. Introduction and Use of the UC‐Denver Veterinary Formulary II. Anesthetic and Analgesic Considerations III. Species Specific Veterinary Formulary 1. Mouse 2. Rat 3. Neonatal Rodent 4. Guinea Pig 5. Chinchilla 6. Gerbil 7. Rabbit 8. Dog 9. Pig 10. Sheep 11. Non‐Pharmaceutical Grade Anesthetics IV. References I. Introduction and Use of the UC‐Denver Formulary Basic Definitions: Anesthesia: central nervous system depression that provides amnesia, unconsciousness and immobility in response to a painful stimulation. Drugs that produce anesthesia may or may not provide analgesia (1, 2). Analgesia: The absence of pain in response to stimulation that would normally be painful. An analgesic drug can provide analgesia by acting at the level of the central nervous system or at the site of inflammation to diminish or block pain signals (1, 2). Sedation: A state of mental calmness, decreased response to environmental stimuli, and muscle relaxation. This state is characterized by suppression of spontaneous movement with maintenance of spinal reflexes (1). Animal anesthesia and analgesia are crucial components of an animal use protocol. This document is provided to aid in the design of an anesthetic and analgesic plan to prevent animal pain whenever possible. However, this document should not be perceived to replace consultation with the university’s veterinary staff. As required by law, the veterinary staff should be consulted to assist in the planning of procedures where anesthetics and analgesics will be used to avoid or minimize discomfort, distress and pain in animals (3, 4). Prior to administration, all use of anesthetics and analgesic are to be approved by the Institutional Animal Care and Use Committee (IACUC). -

Heterodimerization of Μ and Δ Opioid Receptors: a Role in Opiate Synergy
The Journal of Neuroscience, 2000, Vol. 20 RC110 1of5 Heterodimerization of and ␦ Opioid Receptors: A Role in Opiate Synergy I. Gomes, B. A. Jordan, A. Gupta, N. Trapaidze, V. Nagy, and L. A. Devi Departments of Pharmacology and Anesthesiology, New York University School of Medicine, New York, New York 10016 Opiate analgesics are widely used in the treatment of severe -selective ligands results in a significant increase in the bind- pain. Because of their importance in therapy, different strate- ing of a ␦ receptor agonist. This robust increase is also seen in gies have been considered for making opiates more effective SKNSH cells that endogenously express both and ␦ recep- while curbing their liability to be abused. Although most opiates tors. Furthermore, we find that a ␦ receptor antagonist en- exert their analgesic effects primarily via opioid receptors, a hances both the potency and efficacy of the receptor signal- number of studies have shown that ␦ receptor-selective drugs ing; likewise a antagonist enhances the potency and efficacy can enhance their potency. The molecular basis for these find- of the ␦ receptor signaling. A combination of agonists ( and ␦ ings has not been elucidated previously. In the present study, receptor selective) also synergistically binds and potentiates we examined whether heterodimerization of and ␦ receptors signaling by activating the –␦ heterodimer. Taken together, could account for the cross-modulation previously observed these studies show that heterodimers exhibit distinct ligand between these two receptors. We find that co-expression of binding and signaling characteristics. These findings have im- and ␦ receptors in heterologous cells followed by selective portant clinical ramifications and may provide new foundations immunoprecipitation results in the isolation of –␦ het- for more effective therapies. -

Pharmaceutical Appendix to the Tariff Schedule 2
Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. ABACAVIR 136470-78-5 ACIDUM LIDADRONICUM 63132-38-7 ABAFUNGIN 129639-79-8 ACIDUM SALCAPROZICUM 183990-46-7 ABAMECTIN 65195-55-3 ACIDUM SALCLOBUZICUM 387825-03-8 ABANOQUIL 90402-40-7 ACIFRAN 72420-38-3 ABAPERIDONUM 183849-43-6 ACIPIMOX 51037-30-0 ABARELIX 183552-38-7 ACITAZANOLAST 114607-46-4 ABATACEPTUM 332348-12-6 ACITEMATE 101197-99-3 ABCIXIMAB 143653-53-6 ACITRETIN 55079-83-9 ABECARNIL 111841-85-1 ACIVICIN 42228-92-2 ABETIMUSUM 167362-48-3 ACLANTATE 39633-62-0 ABIRATERONE 154229-19-3 ACLARUBICIN 57576-44-0 ABITESARTAN 137882-98-5 ACLATONIUM NAPADISILATE 55077-30-0 ABLUKAST 96566-25-5 ACODAZOLE 79152-85-5 ABRINEURINUM 178535-93-8 ACOLBIFENUM 182167-02-8 ABUNIDAZOLE 91017-58-2 ACONIAZIDE 13410-86-1 ACADESINE 2627-69-2 ACOTIAMIDUM 185106-16-5 ACAMPROSATE 77337-76-9 -

Modulation of Opioid Transport at the Blood-Brain Barrier by Altered ATP-Binding Cassette (ABC) Transporter Expression and Activity
pharmaceutics Review Modulation of Opioid Transport at the Blood-Brain Barrier by Altered ATP-Binding Cassette (ABC) Transporter Expression and Activity Junzhi Yang 1, Bianca G. Reilly 2, Thomas P. Davis 1,2 and Patrick T. Ronaldson 1,2,* 1 Department of Pharmacology and Toxicology, College of Pharmacy, University of Arizona, 1295 N. Martin St., P.O. Box 210207, Tucson, AZ 85721, USA; [email protected] (J.Y.); [email protected] (T.P.D.) 2 Department of Pharmacology, College of Medicine, University of Arizona, 1501 N. Campbell Ave, P.O. Box 245050, Tucson, AZ 85724-5050, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-520-626-2173 Received: 19 September 2018; Accepted: 16 October 2018; Published: 18 October 2018 Abstract: Opioids are highly effective analgesics that have a serious potential for adverse drug reactions and for development of addiction and tolerance. Since the use of opioids has escalated in recent years, it is increasingly important to understand biological mechanisms that can increase the probability of opioid-associated adverse events occurring in patient populations. This is emphasized by the current opioid epidemic in the United States where opioid analgesics are frequently abused and misused. It has been established that the effectiveness of opioids is maximized when these drugs readily access opioid receptors in the central nervous system (CNS). Indeed, opioid delivery to the brain is significantly influenced by the blood-brain barrier (BBB). In particular, ATP-binding cassette (ABC) transporters that are endogenously expressed at the BBB are critical determinants of CNS opioid penetration. In this review, we will discuss current knowledge on the transport of opioid analgesic drugs by ABC transporters at the BBB. -

2017 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 35Th Annual Report
CLINICAL TOXICOLOGY https://doi.org/10.1080/15563650.2018.1533727 NPDS REPORT 2017 2017 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 35th Annual Report David D. Gummin MDa,b, James B. Mowry PharmDc, Daniel A. Spyker PhD, MDd,e, Daniel E. Brooks MDf, Krista M. Osterthaler MPHg and William Banner MD, PhDh aWisconsin Poison Center, Milwaukee, WI, USA; bDepartment of Emergency Medicine, Section of Medical Toxicology, Medical College of Wisconsin, Milwaukee, WI, USA; cIndiana Poison Center, Indiana University Health, Indianapolis, IN, USA; dDepartment of Emergency Medicine, Oregon Poison Center, Oregon Health & Science University, Portland, OR, USA; eDepartment of Biopharmaceutical Sciences, University of California, San Francisco, CA, USA; fDepartment of Medical Toxicology, Banner University Medical Center - Phoenix, Phoenix, AZ, USA; gAmerican Association of Poison Control Centers, Alexandria, VA, USA; hOklahoma Center for Poison and Drug Information, University of Oklahoma College of Pharmacy, Oklahoma City, OK, USA Table of contents Introduction .........................................................................................................1 The NPDS products database........................................................................................ 5 Methods . .........................................................................................................5 Characterization of participating poison centers and population served ................................................. -
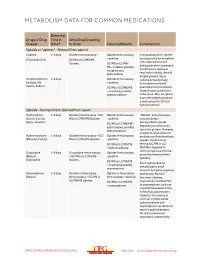
Metabolism Data for Common Medications
METABOLISM DATA FOR COMMON MEDICATIONS Detection Drugs or Drug Time in Urine Drug Screening Classes Urine* to Order Expected Results Consideration Opioids or “opiates” – Natural (from opium) Codeine 1–3 days Opiates Immunoassay + Opiates Immunoassay Immunoassays for “opiates” are responsive for morphine (Tylenol #2/3/4) GC/MS or LC/MS/MS – positive and codeine but do not Opiates GC/MS or LC/MS/ distinguish which is present. MS – codeine, possibly Confirmatory testing is morphine and required to reliably identify hydrocodone drug(s) present. Since Morphine (Avinza, 1–3 days Opiates Immunoassay codeine is metabolized Embeda, MS – positive to morphine and small Contin, Kadian) GC/MS or LC/MS/MS quantities to hydrocodone, – morphine, possibly these drugs may be found hydromorphone in the urine. Also, morphine may metabolize to produce a small amount (<10%) of hydromorphone. Opioids – Semisynthetic (derived from opium Hydrocodone 1–3 days Opiates Immunoassay + GC/ Opiates Immunoassay “Opiates” immunoassays (Lorcet, Lortab, MS or LC/MS/MS Opiates – positive may also detect Norco, Vicodin) semisynthetic opioids GC/MS or LC/MS/MS – depending on their cross- hydrocodone, possibly reactivity pattern. However, hydromorphone a negative result does not Hydromorphone 1–3 days Opiates Immunoassay + GC/ Opiates Immunoassay exclude use of semisynthetic (Dilaudid, Exalgo) MS or LC/MS/MS Opiates –positive opioids. Confirmatory GC/MS or LC/MS/MS testing (GC/MS or LC/ – hydromorphone MS/MS) is required to verify compliance with the Oxycodone 1–3 days Oxycodone Immunoassay Opiates Immunoassay prescribed semisynthetic (Roxicet, + GC/MS or LC/MS/MS –positive opioid(s). OxyContin) Opiates GC/MS or LC/MS/MS Since hydrocodone is – oxycodone possibly metabolized in small oxymorphone amounts to hydromorphone, Oxymorphone 1–3 days Opiates or Oxycodone Opiates or Oxycodone both may be found in (Opana) Immunoassay + GC/MS or Immunoassay – positive the urine. -

ATP, 489 Absolute Configuration Benzomotphans, 204 Levotphanol
Index AIDA, 495 Affinity labeling, analogs of (Cont.) cAMP, 409, 489 motphine,448 ATP, 409, 489 naltrexone, 449 [3H] ATP, 489 norlevotphanol,449 Absolute configuration normetazocine, 181 benzomotphans, 204 norpethidine, 232 levotphanol, 115 oripavine, 453 methadone and analogs, 316 oxymotphone, 449 motphine, 86 K-Agonists, 179,405,434 phenoperidine, 234 Aid in Interactive Drug Analysis, 495 piperazine derivatives, 399 [L-Ala2] dermotphin, 363 prodines and analogs, 272 [D-Ala, D-Leu] enkephalin (DADL), 68, 344 sinomenine, 28, 115 [D-Ala2 , Bugs] enkephalinamide, 347, 447 viminol, 400 [D-Ala2, Met'] enkephalinamide, 337, 346, Ac 61-91,360 371,489 Acetylcholine, 5, 407 [D-Ala2]leu-enkephalin, 344, 346, 348 Acetylcholine analogs, 186, 191 [D-Ala2] met-enkephalin, 348 l-Acetylcodeine, 32 [D-Ala2] enkephalins, 347 Acetylmethadols (a and (3) Alfentanil, 296 maintenance of addicts by a-isomer, 304, 309 (±)-I1(3-Alkylbenzomotphans, 167, 170 metabolism, 309 11(3-Alkylbenzomotphans, 204 N-allyl and N-CPM analogs, 310, 431 7-Alkylisomotphinans, 146 stereochemistry, 323 N-Alkylnorketobemidones, 431 synthesis, 309 N-Alkylnorpethidines, 233 X-ray crystallography, 327 N-Allylnormetazocine, 420 6-Acetylmotphine, receptor binding, 27 N-Allylnormotphine, 405 Acetylnormethadol, 323 N-Allylnorpethidine, 233 8(3-Acyldihydrocodeinones, 52 3-Allylprodines (a and (3), 256 14-Acyl-4,5-epoxymotphinans, 58 'H-NMR and stereochemistry, 256 7-Acylhydromotphones, 128 X-ray crystallography, 256 Addiction, 4 N-Allylnormetazocine, 420 Adenylate cyclase, 6, 409, 413, 424, -

Symposium Iv. Discriminative Stimulus Effects
Life Sciences, Vol. 28, pp. 1571-1584 Pergamon Press Printed in the U.S.A. MINI - SYMPOSIUM IV. DISCRIMINATIVE STIMULUS EFFECTS OF NARCOTICS: EVIDENCE FOR MULTIPLE RECEPTOR-MEDIATED ACTIONS Seymore Herling and James H. Woods Departments of Pharmacology and Psychology University of Michigan Ann Arbor, Michigan q8109 The different pharmacological syndromes produced by morphine and related drugs in the chronic spinal dog led Martin and his colleagues (1,2) to suggest that these drugs exert their agonist actions 0y interacting with three distinct receptors (~,K, and e). Morphine was hypothesized to be an agonist for the p receptor, ketazocine (ketocyclazocine) was an agonist for the K receptor, and SKF-10,0q7 was an agonist for the ~ receptor. The effects of these three drugs in the chronic spinal dog were reversed by the narcotic antagonist, naltrexone, indicating that the effects of these drugs are narcotic agonist effects (I). In additlon to the different effects of these narcotics in the non- dependent chronic spinal dog, the effects of morphine, ketazocine, and SKF-IO,047 in several other behavioral and physiological preparations are consistent with the concept of multiple receptors. For example, while ketazocine and ethylketazocine, like morphine, produce analgesia, these compounds, unlike morphine, do not suppress signs of narcotic abstinence in the morphine-dependent rhesus monkey or morphine-dependent chronic spinal dog (1-5). Further, the characteristics of ketazocine withdrawal and antagonist- precipitated abstinence syndromes, although similar to those of cyclazocine, are quailtativeiy different from those of morphine (1,2). In rhesus monkeys, ketazocine, ethylketazocine, and SKF-10,047 maintain lever pressing at rates comparable to or below those maintained by saline, and well below response rates maintained by codeine or morphine (5,6), suggesting that the former set of drugs have limited reinforcing effect. -

Summary of Synthetic Opioid Testing Within the Department of Defense
SUMMARY OF SYNTHETIC OPIOID TESTING WITHIN THE DEPARTMENT OF DEFENSE Tom Martin, Ph.D. Liaison to Director, DoD Drug Testing and Program Policy PERSONNEL AND READINESS PERSONNEL AND READINESS Drug Demand Reduction Program Mission and Scope Mission: Enable operational readiness, safety, and security of the Total Force by deterring illicit and prescription drug abuse through robust and dynamic drug testing; emerging drug threat surveillance; prevention, education, and outreach efforts; and development of new testing procedures. Scope: All DoD components and DoD civilians in testing designated positions (TDPs) Policies: DODI 1010.01 “Military Personnel Drug Abuse Testing Program (MPDATP)” DODI 1010.09 “DoD Civilian Employee Drug-Free Workplace Program” DODI 1010.16 “Technical Procedures for the Military Personnel Drug Abuse Testing Program (MPDATP)” PERSONNEL AND READINESS DDRP Driving Factors • Drug abuse in the general U.S. 18-25 year old male group is estimated to be 17-20%– the population from which the Service recruits their enlisted personnel • Before DoD instituted drug testing among Service personnel, drug use was a significant recurring problem – Vietnam (estimated over 5% of returning service members addicted to heroin) – 1981 CVN Nimitz aviation mishap – 14 killed, 48 injured, 7 aircraft destroyed, 11 aircraft damaged, $150M in damages, six deceased with detectable levels of marijuana • Notable increase in abuse/misuse of prescription pain medications • Personnel abusing illicit drugs or prescription medications are a safety hazard