A Laboratory Guide to Clinical Hematology
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Hemolytic Disease of the Newborn
Intensive Care Nursery House Staff Manual Hemolytic Disease of the Newborn INTRODUCTION and DEFINITION: Hemolytic Disease of the Newborn (HDN), also known as erythroblastosis fetalis, isoimmunization, or blood group incompatibility, occurs when fetal red blood cells (RBCs), which possess an antigen that the mother lacks, cross the placenta into the maternal circulation, where they stimulate antibody production. The antibodies return to the fetal circulation and result in RBC destruction. DIFFERENTIAL DIAGNOSIS of hemolytic anemia in a newborn infant: -Isoimmunization -RBC enzyme disorders (e.g., G6PD, pyruvate kinase deficiency) -Hemoglobin synthesis disorders (e.g., alpha-thalassemias) -RBC membrane abnormalities (e.g., hereditary spherocytosis, elliptocytosis) -Hemangiomas (Kasabach Merritt syndrome) -Acquired conditions, such as sepsis, infections with TORCH or Parvovirus B19 (anemia due to RBC aplasia) and hemolysis secondary to drugs. ISOIMMUNIZATION A. Rh disease (Rh = Rhesus factor) (1) Genetics: Rh positive (+) denotes presence of D antigen. The number of antigenic sites on RBCs varies with genotype. Prevalence of genotype varies with the population. Rh negative (d/d) individuals comprise 15% of Caucasians, 5.5% of African Americans, and <1% of Asians. A sensitized Rh negative mother produces anti-Rh IgG antibodies that cross the placenta. Risk factors for antibody production include 2nd (or later) pregnancies*, maternal toxemia, paternal zygosity (D/D rather than D/d), feto-maternal compatibility in ABO system and antigen load. (2) Clinical presentation of HDN varies from mild jaundice and anemia to hydrops fetalis (with ascites, pleural and pericardial effusions). Because the placenta clears bilirubin, the chief risk to the fetus is anemia. Extramedullary hematopoiesis (due to anemia) results in hepatosplenomegaly. -

Evaluation of Anemia Survey (NHANES III) Data- 9 10-28% of Patients Over 65 Years Are Anemic Mark Wurster, M.D., F.A.C.P
Anemia - Definition • National Health and Nutrition Examination Evaluation of Anemia Survey (NHANES III) data- 9 10-28% of patients over 65 years are anemic Mark Wurster, M.D., F.A.C.P. 9 One third of these are due to iron, folate, B12 The Ohio State University deficiency alone or in combination 9 One third are due to renal disease, or other chronic inflammatory response 9 One third are due to various primary marrow disorders, malignancies or other disorders Anemia Anemia - Definition Classification Schemes • A simplified approach to anemia, • Most common hematologic disorder emphasizing information already included • Decrease from normal levels of Hgb, Hct, RBC: in the CBC: 9 FlFemales – MHb14/dlMean Hgb = 14 g/dl; -2SD = 12 g /dl • Mean Cellular Volume (MCV) 9 Males – Mean Hgb = 15.5 g/dl; -2SD = 13.5 g/dl • Red Cell Distribution Width (RDW) • Caveat – Anemia is a syndrome, not a disease. • Retic count An abnormal Hgb or Hct should ALWAYS be investigated if confirmed on repeat testing. 1 Anemia Anemia Classification Schemes Classification Schemes • Mean Cellular Volume (MCV) • Red blood cell Distribution Width (RDW) • Decreased MCV (microcytic); < 80 fL 9 A numerical expression of • Normal MCV (normocytic); 80 – 99 fL anisocytosis, or variation in RBC size • Increased MCV (macrocytic); > 100 fL Anemia Anemia Classification Schemes Classification Schemes • Red blood cell Distribution Width (RDW) 9 Normal RDW - representing a uniform population • Red blood cell Distribution Width (RDW) of RBCs with respect to size (actually the standard deviation of red blood cell volume divided by the mean volume) 9 Normal; < or = to app. -

Reactive Plasmacytosis in a Patient of Acute Myeloid Leukemia at Initial
Open Journal of Clinical & Medical Volume 7 (2021) Case Reports Issue 01 ISSN: 2379-1039 Reactive plasmacytosis in a patient of acute myeloid leukemia at initial presentation – A rare occurrence Sundas Ali; Shahzad Ali Jiskani*; Samina Tufail Amanat; Aliena Sohail *Corresponding Author: Shahzad Ali Jiskani Department of Pathology, Pakistan Institute of Medical Sciences, Islamabad. Email: [email protected] Abstract An increased proliferation of plasma cells has been seen very rarely in patients of Acute Myeloid Leukemia (AML) at the time of initial presentation. In this report, we describe a 64-year old case of AML with reactive plasmacytosis at the time of diagnosis. It has been suggested that paraneoplastic IL-6 production by leuke- mic blasts may be responsible for growth stimulation of plasma cells in marrow. Keywords Acute myeloid leukemia; reactive plasmacytosis; paraneoplastic IL-6. Background The presence of reactive plasmacytosis in patients with acute myeloid leukemia has been shown after chemotherapy. However, it is a rare occurrence in a newly diagnosed AML case. Our patient presen- ted with a moderate proliferation of plasma cells in the bone marrow, along with morphological features consistent with the diagnosis of acute myeloid leukemia with maturation. A careful investigative approach is required to resolve this diagnostic dilemma [1,2]. Case presentation This 64-year old male patient presented to our hospital with complaints of fever, productive cough, generalized weakness and undocumented weight loss for the last one month, and epistaxis and hematuria for the last 5 days. He was a known case of HCV taking no treatment. Physical examination revealed only pallor and mild jaundice. -

Modelling of Red Blood Cell Morphological and Deformability Changes During In-Vitro Storage
applied sciences Article Modelling of Red Blood Cell Morphological and Deformability Changes during In-Vitro Storage Nadeeshani Geekiyanage 1 , Emilie Sauret 1,*, Suvash Saha 2 , Robert Flower 3 and YuanTong Gu 1 1 School of Mechanical, Medical and Process Engineering, Science and Engineering Faculty, Queensland University of Technology (QUT), Brisbane City, QLD 4000, Australia; [email protected] (N.G.); [email protected] (Y.G.) 2 School of Mechanical and Mechatronic Engineering, University of Technology Sydney (UTS), Ultimo, NSW 2007, Australia; [email protected] 3 Research and Development, Australian Red Cross Lifeblood, Kelvin Grove, QLD 4059, Australia; [email protected] * Correspondence: [email protected] Received: 28 February 2020; Accepted: 27 April 2020; Published: 4 May 2020 Featured Application: Red blood cell (RBC) storage lesion is a critical issue facing transfusion treatments, and significant changes in RBC morphology and deformability are observed due to the storage lesion. RBCs require high deformability to sustain in-vivo circulation, and impaired deformability leads to several post-transfusion adverse outcomes. Therefore, improved understanding of the interrelation between the morphological and deformability changes and the quality and viability of the stored RBCs is essential to prevent or reduce the transfusion related adverse outcomes. To support this requisite, the influence on RBC deformability due to several aspects of the storage lesion, namely, the changes in cell morphology, surface area and volume, RBC membrane biomechanics, and cytoskeletal structural integrity are explored numerically in this study. Abstract: Storage lesion is a critical issue facing transfusion treatments, and it adversely affects the quality and viability of stored red blood cells (RBCs). -

Age-Related Features and Pathology of Blood in Children
MINISTRY OF PUBLIC HEALTH OF UKRAINE HIGHER STATE EDUCATIONAL ESTABLISHMENT OF UKRAINE «UKRAINIAN MEDICAL STOMATOLOGICAL ACADEMY» V.I. POKHYLKO, S.M. TSVIRENKO, YU.V. LYSANETS AGE-RELATED FEATURES AND PATHOLOGY OF BLOOD IN CHILDREN MANUAL FOR STUDENTS OF HIGHER MEDICAL EDUCATIONAL INSTITUTIONS OF THE III-IV ACCREDITATION LEVELS Poltava 2017 МІНІСТЕРСТВО ОХОРОНИ ЗДОРОВ’Я УКРАЇНИ ВИЩИЙ ДЕРЖАВНИЙ НАВЧАЛЬНИЙ ЗАКЛАД УКРАЇНИ «УКРАЇНСЬКА МЕДИЧНА СТОМАТОЛОГІЧНА АКАДЕМІЯ» ПОХИЛЬКО В.І., ЦВІРЕНКО С.М., ЛИСАНЕЦЬ Ю.В. ВІКОВІ ОСОБЛИВОСТІ ТА ПАТОЛОГІЯ КРОВІ У ДІТЕЙ НАВЧАЛЬНИЙ ПОСІБНИК ДЛЯ СТУДЕНТІВ ВИЩИХ МЕДИЧНИХ НАВЧАЛЬНИХ ЗАКЛАДІВ III-IV РІВНІВ АКРЕДИТАЦІЇ Полтава 2017 2 UDC: 616+616.15]-053.2(075.8) ВВС: 57.33я73 The manual highlights the issues of embryogenesis, age-related features, semiotics of lesion, examination methods and diseases of hemic system in children. The manual is intended for students of higher educational institutions of III-IV accreditation levels, and can be used by medical interns and primary care doctors. Authors: Doctor of Medical Sciences, Professor of the Department of Pediatrics No.1 with Propedeutics and Neonatology V.I. Pokhylko Candidate of Medical Sciences, Acting Head of the Department of Pediatrics No.1 with Propedeutics and Neonatology S.M. Tsvirenko Candidate of Philological Sciences, Senior Lecturer of the Department of Foreign Languages with Latin and Medical Terminology Yu.V. Lysanets Reviewers: O.S. Yablon’ ― Doctor of Medical Sciences, Professor, Head of the Department of Pediatrics No.1, Vinnytsya National M.I. Pirogov Memorial Medical University of Ministry of Public Health of Ukraine. M.O. Honchar ― Doctor of Medical Sciences, Professor, Head of the Department of Pediatrics and Neonatology No.1, Kharkiv National Medical University. -

Anemia and the QT Interval in Hypertensive Patients
2084 International Journal of Collaborative Research on Internal Medicine & Public Health Anemia and the QT interval in hypertensive patients Ioana Mozos 1*, Corina Serban 2, Rodica Mihaescu 3 1 Department of Functional Sciences, “Victor Babes” University of Medicine and Pharmacy, Timisoara, Romania 2 Department of Functional Sciences, “Victor Babes” University of Medicine and Pharmacy, Timisoara, Romania 3 1st Department of Internal Medicine, “Victor Babes” University of Medicine and Pharmacy, Timisoara, Romania * Corresponding Author ; Email: [email protected] Abstract Introduction: A prolonged ECG QT interval duration and an increased QT dispersion (QTd) are predictors of sudden cardiac death. Anemia is known as a marker of adverse outcome in cardiovascular disease. Objective: The aim was to assess the relationship between anemia and QT intervals in hypertensive patients. Method: A total of 72 hypertensive patients underwent standard 12-lead ECG. QT intervals and QT dispersions were manually measured. Complete blood count was also assessed. Result: Linear regression analysis revealed significant associations between prolonged QTc and increased QTd and anemia and macrocytosis, respectively. Multiple regression analysis revealed a significant association between red cell distribution width (RDW) >15% and prolonged heart rate corrected maximal QT interval duration (QTc) and QT interval in lead DII (QTIIc). The most sensitive and specific predictor of prolonged QTc and QTIIc was anisocytosis. Anemia was the most sensitive predictor of -
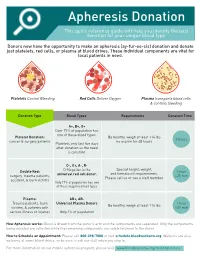
Apheresis Donation This Quick Reference Guide Will Help You Identify the Best Donation for Your Unique Blood Type
Apheresis Donation This quick reference guide will help you identify the best donation for your unique blood type. Donors now have the opportunity to make an apheresis (ay-fur-ee-sis) donation and donate just platelets, red cells, or plasma at blood drives. These individual components are vital for local patients in need. Platelets Control Bleeding Red Cells Deliver Oxygen Plasma transports blood cells & controls bleeding Donation Type Blood Types Requirements Donation Time A+, B+, O+ Over 75% of population has one of these blood types. Platelet Donation: Be healthy, weigh at least 114 lbs 2 hours cancer & surgery patients no aspirin for 48 hours Platelets only last five days after donation so the need is constant. O-, O+, A-, B- Special height, weight, Double Red: O-Negative is the 1 hour and hematocrit requirements. surgery, trauma patients, universal red cell donor. +25 min Please call us or see a staff member accident, & burn victims Only 17% of population has one of these negative blood types Plasma: AB+, AB- Trauma patients, burn Universal Plasma Donors 1 hour Be healthy, weigh at least 114 lbs victims, & patients with +30 min serious illness or injuries Only 4% of population How Apheresis works: Blood is drawn from the donor’s arm and the components are separated. Only the components being donated are collected while the remaining components are safely returned to the donor How to Schedule an Appointment: Please call 800-398-7888 or visit schedule.bloodworksnw.org. Walk-ins are also welcome at some blood drives, so be sure to ask our staff when you stop in. -

The Complete Blood Count Sample Report 1
THE COMPLETE BLOOD COUNT SAMPLE REPORT 1. Name and address of the lab where the test was performed. Tests may be run in a physician office lab, a lab located in a Different laboratories generate reports that can vary greatly in appearance and in the order and kind of The Complete Blood Count Sample Report clinic or hospital, and/or samples may be sent to a reference information included. This is one example of what a lab report for a Complete Blood Count may look like. laboratory for analysis. NamesDifferent and laboratories places used generate have been reports made that up can for vary illustrative greatly inpurposes appearance only. andThe innumbered the order keyand tokind the of right 2. Date this copy of the report was printed. This date may be explainsinformation a few included. of the reportThis is elements.one exampl e of what a lab report for a Complete Blood Count may look like. different than the date the results were generated, especially Names and places used have been made up for illustrative purposes only. Point your cursor at a number on cumulative reports (those that include results of several to learn about the different report elements. different tests run on different days). 3. Patient name or identifier. Links results to the correct person. 1 University Medical Center, Dept. of Pathology Report Date/Time: 123 University Way, City, ST 12345 02/10/2014 16:40 2 4. Patient identifier and identification number. Links results to the correct person. 3 Name: Doe, John Q. Age/Sex: 73/M DOB: 01/01/1941 5. -

Erythrocytes: Overview, Morphology, Quantity by AH Rebar Et
In: A Guide to Hematology in Dogs and Cats, Rebar A.H., MacWilliams P.S., Feldman B.F., Metzger F.L., Pollock R.V.H. and Roche J. (Eds.). Publisher: Teton NewMedia, Jackson WY (www.veterinarywire.com). Internet Publisher: International Veterinary Information Service, Ithaca NY (www.ivis.org), 8-Feb-2005; A3304.0205 Erythrocytes: Overview, Morphology, Quantity A.H. Rebar1, P.S. MacWilliams2, B.F. Feldman 3, F.L. Metzger 4, R.V.H. Pollock 5 and J. Roche 6 1Dept of Veterinary Pathobiology, School of Veterinary Medicine, Purdue University, IN,USA. 2Dept of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin, WI, USA. 3Dept of Biomedical Sciences & Pathobiology, VA-MD - Regional College of Veterinary Medicine, Virginia Tech, VA, USA. 4Metzger Animal Hospital,State College,PA, USA. 5Fort Hill Company, Montchanin, DE, USA. 6 Hematology Systems, IDEXX Laboratories, Westbrook, ME, USA. Overview Production Red blood cells (RBC) are produced in the bone marrow. Numbers of circulating RBCs are affected by changes in plasma volume, rate of RBC destruction or loss, splenic contraction, erythropoietin (EPO) secretion, and the rate of bone marrow production. A normal PCV is maintained by an endocrine loop that involves generation and release of erythropoietin (EPO) from the kidney in response to renal hypoxia. Erythropoietin stimulates platelet production as well as red cell production. However, erythropoietin does not stimulate white blood cell (WBC) production. Erythropoiesis and RBC numbers are also affected by hormones from the adrenal cortex, thyroid, ovary, testis, and anterior pituitary. Destruction Red cells have a finite circulating lifespan. In dogs, the average normal red cell circulates approximately 100 days. -

Patient Information Leaflet – Plasma Exchange Procedure
Therapeutic Apheresis Services Patient Information Leaflet – Plasma Exchange Procedure Introduction Antibodies, which normally help to protect you from infection, can begin to attack your own This leaflet has been written to give patients healthy cells, or an over production of proteins can information about plasma exchange (sometimes cause your blood to become thicker and slow down called plasmapheresis). If you would like any more the blood flow throughout your body. A plasma information or have any questions, please ask the exchange can help improve your symptoms, doctors and nurses involved in your treatment at although this may not happen immediately. the NHS Blood and Transplant (NHSBT) Therapeutic Apheresis Services Unit. Although plasma exchange may help with symptoms, it will not normally cure your condition When you have considered the information given as it does not switch off the production of the in this leaflet, and after we have discussed the harmful antibodies or proteins. It is likely that procedure and its possible risks with you, we will this procedure will form only one part of your ask you to sign a consent form to indicate that you treatment. are happy for the procedure to go ahead. Before any further procedures we will again check that you are happy to proceed. How do we perform Plasma Exchange? What is a plasma exchange? Plasma exchange is performed using a machine Blood is made up of red cells, white cells and called a Blood Cell Separator which can separate platelets which are carried around in fluid called blood into its various parts. The machine separates plasma. -

We Get Biased on Things That Make Sense
DIALOGUE AND DISCUSSION We Get Biased on Things That Make Sense MERIH T. TESFAZGHI When I was teaching third year medical students, and bench- reaction might be characterized by a marked left shift lecturing clinical laboratory science students, I emphasized which may display a range of immature cells that are also the importance of clinical details and the history of patients seen in CML. Severe left shift especially in the absence of in laboratory test processing, especially in the evaluation of evident infection (or other underlying diseases) may peripheral blood films. During those times, one of my strongly suggest direct bone marrow involvement. In students stated, “but including details of patients might such scenario, clinical details of patients such as absence somehow affect the decision of the reviewer, and may lead to or presence of enlargement of extramedullar organs is Downloaded from a bias.” I responded that we are biased on things that make highly sought. sense. This means that we decide based on the evidence we see from a microscopy evaluation in the light of the clinical Ø The coexistence of multiple conditions in a given detail and history. How do clinical details and history help us patient. One condition may mask the presence of the reach decisions? other condition. For example, the coexistence of megaloblastic anemia with iron deficiency anemia. http://hwmaint.clsjournal.ascls.org/ If specimens are the “in vitro ambassadors” of patients to the Usually, megaloblastic anemia is characterized by laboratory, then properly completed request forms are their increased size of red blood cells (MCV), but when “credentials”. -

Evaluation of Complete Blood Count Parameters for Diagnosis In
Original Investigation / Özgün Araştırma DOI: 10.5578/ced.68886 • J Pediatr Inf 2020;14(2):e55-e62 Evaluation of Complete Blood Count Parameters for Diagnosis in Children with Sepsis in the Pediatric Intensive Care Unit Pediatrik Yoğun Bakım Ünitesinde Sepsisli Çocuklarda Tanı İçin Tam Kan Sayımı Parametrelerinin Değerlendirilmesi Fatih Aygün1(İD), Cansu Durak1(İD), Fatih Varol1(İD), Haluk Çokuğraş2(İD), Yıldız Camcıoğlu2(İD), Halit Çam1(İD) 1 Department of Pediatric Intensive Care, Istanbul University School of Cerrahpasa Medicine, Istanbul, Turkey 2 Department of Pediatric Infectious Diseases, Istanbul University School of Cerrahpasa Medicine, Istanbul, Turkey Cite this article as: Aygün F, Durak C, Varol F, Çokuğraş H, Camcıoğlu Y, Çam H. Evaluation of complete blood count parameters for diagnosis in children with sepsis in the pediatric intensive care unit. J Pediatr Inf 2020;14(2):e55-e62. Abstract Öz Objective: Early diagnosis of sepsis is important for effective treatment Giriş: Erken tanı sepsiste etkili tedavi ve iyi prognoz için önemlidir. C-re- and improved prognosis. C-reactive protein (CRP) and procalcitonin aktif protein (CRP) ve prokalsitonin (PKT) sepsiste en sık kullanılan bi- (PCT) are the most commonly used biomarkers for sepsis. However, their yobelirteçlerdir. Fakat rutinde kullanımı maliyet etkin değildir. Tam kan routine usage is not cost-effective. Complete Blood Count (CBC) param- sayımı (TKS) parametrelerinden eritrosit dağılım genişliği (EDG), nötrofil eters including red cell distribution width (RDW), neutrophil to lympho- lenfosit oranı (NLO), trombosit lenfosit oranı (TLO), ortalama trombosit cyte ratios (NLR), platelet to lymphocyte ratios (PLR), mean platelet vol- hacmi (OTH) basit ve kolay olarak hesaplanmaktadır. Bu çalışmanın ama- ume (MPV), and hemoglobin are simple and easily calculated.