Evidence for Nonacetylcholinesterase Targets of Organophosphorus Nerve Agent: Supersensitivity of Acetylcholinesterase Knockout Mouse to VX Lethality
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Review of the Mammalian Toxicology
REVIEW OF THE MAMMALIAN TOXICOLOGY AND METABOLISM/TOXICOKINETICS OF FENTHION The APVMA Review of Fenthion © Australian Pesticides and Veterinary Medicines Authority 2012 ISBN 978-0-9873591-5-5 (electronic) This document was originally published in 2005 as part of the preliminary review findings report for Part 1 of the fenthion review. It was subsequently updated in 2007, and revised in 2008. Ownership of intellectual property rights in this publication Unless otherwise noted, copyright (and any other intellectual property rights, if any) in this publication is owned by the Australian Pesticides and Veterinary Medicines Authority (APVMA). Creative Commons licence With the exception of the Coat of Arms, this publication is licensed under a Creative Commons Attribution 3.0 Australia Licence. This is a standard form agreement that allows you to copy, distribute, transmit and adapt this publication provided that you attribute the work. A summary of the licence terms is available from www.creativecommons.org/licenses/by/3.0/au/deed.en. The full licence terms are available from www.creativecommons.org/licenses/by/3.0/au/legalcode. The APVMA’s preference is that you attribute this publication (and any approved material sourced from it) using the following wording: Source: Licensed from the Australian Pesticides and Veterinary Medicines Authority (APVMA) under a Creative Commons Attribution 3.0 Australia Licence. This report was prepared for the APVMA by the Department of Health and Aging Office of Chemical Safety. In referencing this document the Department of Health and Aging Office of Chemical Safety should be cited as the author and the Australian Pesticides and Veterinary Medicines Authority as the publisher and copyright owner. -

Ortho-Cresyl-Phosphate Poisoning
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.25.3.234 on 1 August 1962. Downloaded from J. Neurol. Neurosurg. Psychiat., 1962, 25, 234 Toxic polyneuritis in Bombay due to ortho-cresyl-phosphate poisoning D. D. VORA, DARAB K. DASTUR, BEATRIZ M. BRAGANCA, L. M. PARIHAR, C. G. S. IYER, R. B. FONDEKAR, AND K. PRABHAKARAN From Lokmanya Tilak Municipal General Hospital, Sion, Neurology Unit, Indian Council of Medical Research, and Department of Enzyme Chemistry, Indian Cancer Research Centre, Bombay Since 1930 poisoning with ortho-cresyl-phosphate purchased food from the same grocer in their neigh- has been recognized as a cause of peripheral poly- bourhood. He also mentioned that others staying in neuritis (Smith, Elvove, and Frazier, 1930). Ortho- the same locality and consuming mustard oil, but cresyl-phosphate (O.C.P.) is widely used as a purchasing their food from other grocers, were not plasticiser and in the production of heat-stable affected. The examination of the other affected per- lubricating oils. Many cases of industrial poisoning sons revealed identical histories and clinical pictures. with O.C.P. have been reported since then but as a In this group there was no history of gastro-enteritis result of strict precautionary measures it has now or of febrile illness preceding the paralysis. These become a rarity. However, the occasional contamina- patients also asserted that all those who were buying is not uncommon. their food from the same grocer but who were not tion of food with O.C.P. Protected by copyright. Poisoning with O.C.P. -

The Effects of Occupational Exposure to Chlorpyrifos on the Peripheral
201 Occup Environ Med: first published as 10.1136/oem.2003.008847 on 25 February 2004. Downloaded from ORIGINAL ARTICLE The effects of occupational exposure to chlorpyrifos on the peripheral nervous system: a prospective cohort study J W Albers, D H Garabrant, S J Schweitzer, R P Garrison, R J Richardson, S Berent ............................................................................................................................... Occup Environ Med 2004;61:201–211. doi: 10.1136/oem.2003.008847 Aims: To determine whether chronic occupational exposure to chlorpyrifos at levels associated with various aspects of manufacturing produced a clinically evident or subclinical peripheral neuropathy. Methods: Clinical and quantitative nerve conduction study (NCS) examinations were performed on two occasions on chlorpyrifos manufacturing workers who had measurable chlorpyrifos exposure and a referent group. Baseline evaluations were performed on 53 of 66 eligible chlorpyrifos subjects and on 60 of 74 eligible referent subjects; one-year evaluations were completed on 111 of the 113 subjects evaluated at baseline. Results: Chlorpyrifos and referent groups differed significantly in measures of 3,5,6 trichloro-2-pyridinol excretion and plasma butyrylcholinesterase (BuChE) activity, indicating substantially higher exposures See end of article for authors’ affiliations among chlorpyrifos subjects. Few subjects had clinically important neurological symptoms or signs. NCS ....................... results were comparable to control values, and there were no significant group differences in NCS results at baseline, one year, or change over one year. No chlorpyrifos subject fulfilled conventional criteria for Correspondence to: Dr J W Albers, Department confirmed peripheral neuropathy at baseline or one-year examinations. The odds ratios for developing of Neurology, 1C325/ any diagnosable level of peripheral neuropathy among the chlorpyrifos subjects was not increased at 0032 University Hospital, baseline or at one year compared to referents at baseline. -

Enzymatic Degradation of Organophosphorus Pesticides and Nerve Agents by EC: 3.1.8.2
catalysts Review Enzymatic Degradation of Organophosphorus Pesticides and Nerve Agents by EC: 3.1.8.2 Marek Matula 1, Tomas Kucera 1 , Ondrej Soukup 1,2 and Jaroslav Pejchal 1,* 1 Department of Toxicology and Military Pharmacy, Faculty of Military Health Sciences, University of Defence, Trebesska 1575, 500 01 Hradec Kralove, Czech Republic; [email protected] (M.M.); [email protected] (T.K.); [email protected] (O.S.) 2 Biomedical Research Center, University Hospital Hradec Kralove, Sokolovska 581, 500 05 Hradec Kralove, Czech Republic * Correspondence: [email protected] Received: 26 October 2020; Accepted: 20 November 2020; Published: 24 November 2020 Abstract: The organophosphorus substances, including pesticides and nerve agents (NAs), represent highly toxic compounds. Standard decontamination procedures place a heavy burden on the environment. Given their continued utilization or existence, considerable efforts are being made to develop environmentally friendly methods of decontamination and medical countermeasures against their intoxication. Enzymes can offer both environmental and medical applications. One of the most promising enzymes cleaving organophosphorus compounds is the enzyme with enzyme commission number (EC): 3.1.8.2, called diisopropyl fluorophosphatase (DFPase) or organophosphorus acid anhydrolase from Loligo Vulgaris or Alteromonas sp. JD6.5, respectively. Structure, mechanisms of action and substrate profiles are described for both enzymes. Wild-type (WT) enzymes have a catalytic activity against organophosphorus compounds, including G-type nerve agents. Their stereochemical preference aims their activity towards less toxic enantiomers of the chiral phosphorus center found in most chemical warfare agents. Site-direct mutagenesis has systematically improved the active site of the enzyme. These efforts have resulted in the improvement of catalytic activity and have led to the identification of variants that are more effective at detoxifying both G-type and V-type nerve agents. -

Background Information for Chlorpyrifos
64 Appendix A: Background Information for Chlorpyrifos Chlorpyrifos is an organophosphorus insecticide. It belongs to the phosphorothioate (also called phosphorothionate) group, composed of organophosphorus compounds that contain the P=S substructure. Other organophosphorus insecticides in this group include diazinon, methyl parathion, parathion, fenthion, and fenitrothion. These compounds require metabolic activation to their oxon analogs (compounds in which the =S is replaced by =O) for anticholinesterase activity. The structures of chlorpyrifos and its toxic metabolite, chlorpyrifos oxon, are provided in Appendix D. As of 2000, chlorpyrifos was one of the most widely used organophosphorus insecticides in the United States, for both agricultural and residential purposes. Registered uses included food crops, turf and ornamental plants, indoor pest control (including crack and crevice treatment), termite control, mosquito control, and pet collars. It was registered for use in a wide variety of buildings, including residential, commercial, schools, daycare centers, restaurants, hospitals, hotels, and food manufacturing plants (EPA 2000c). Many uses of chlorpyrifos are being phased out (see Section A.4). A study of urinary pesticide metabolites during the third trimester in 386 pregnant women from East Harlem indicated that exposure to chlorpyrifos was prevalent (42% of the women had detectable levels of the chlorpyrifos metabolite), was higher than the median in NHANES III, did not show seasonal variation, and did not change during the -

Qsar Analysis of the Chemical Hydrolysis of Organophosphorus Pesticides in Natural Waters
QSAR ANALYSIS OF THE CHEMICAL HYDROLYSIS OF ORGANOPHOSPHORUS PESTICIDES IN NATURAL WATERS. by Kenneth K. Tanji Principal Investigator and Jonathan 1. Sullivan Graduate Research Assistant Department of Land, Air and Water Resources University of California, Davis Technical Completion Report Project Number W-843 August, 1995 University of California Water Resource Center The research leading to this report was supported by the University of California Water Resource Center as part of Water Resource Center Project W-843. Table of Contents Page Abstract 2 Problem and Research Objectives 3 Introduction 5 Theoretical Background 6 QSAR Methodology 7 Molecular Connectivity Theory 8 Organophosphorus Pesticides 12 Experimental Determination of Rates 15 Results and Discussion 17 Principal Findings and Significance 19 References 34 List of Tables Page Table 1. Statistical relationship between OP pesticides and first-order MC/'s. 30 Table 2. Inherent conditions of waters used in experimental work. 16 Table 3. Estimated half-lives for organophosphorus esters derived from model. 31 Table 4. Half-lives and first-order MCI' sfor model calibration data set. 31 Table 5. Experimental kinetic data for validation set compounds, Sacramento. 33 List of Figures Page Figure 1. Essential Features OfQSAR Modeling Methodology. 21 Figure 2. Regression plot for In hydrolysis rate vs. 1st order MCl' s. 22 Figure 3. a 3-D molecular model, a line-segment model and a graphical model. 23 Figure 4. Molecular connectivity index suborders. 24 Figure 5. Chlorpyrifos and its fourteen fourth order path/cluster fragments. 25 Figure 6. Abridged MClndex output. 26 Figure 7. Parent acids of most common organophosphorus pesticides. 12 Figure 8. -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination -

Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 Theinternational Programme on Chemical Safety (IPCS) Was Established in 1980
The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 cation Hazard of Pesticides by and Guidelines to Classi The WHO Recommended Classi The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 TheInternational Programme on Chemical Safety (IPCS) was established in 1980. The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. This publication was developed in the IOMC context. The contents do not necessarily reflect the views or stated policies of individual IOMC Participating Organizations. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase international coordination in the field of chemical safety. The Participating Organizations are: FAO, ILO, UNDP, UNEP, UNIDO, UNITAR, WHO, World Bank and OECD. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment. WHO recommended classification of pesticides by hazard and guidelines to classification, 2019 edition ISBN 978-92-4-000566-2 (electronic version) ISBN 978-92-4-000567-9 (print version) ISSN 1684-1042 © World Health Organization 2020 Some rights reserved. -

Carbaryl Human Health and Ecological Risk Assessment Revised Final Report
SERA TR-052-01-05a Carbaryl Human Health and Ecological Risk Assessment Revised Final Report Submitted to: Paul Mistretta, COR USDA/Forest Service, Southern Region 1720 Peachtree RD, NW Atlanta, Georgia 30309 USDA Forest Service Contract: AG-3187-C-06-0010 USDA Forest Order Number: AG-43ZP-D-06-0009 SERA Internal Task No. 52-01 Submitted by: Patrick R. Durkin and Cynthia King Syracuse Environmental Research Associates, Inc. 5100 Highbridge St., 42C Fayetteville, New York 13066-0950 Fax: (315) 637-0445 E-Mail: [email protected] Home Page: www.sera-inc.com February 9, 2008 Table of Contents Table of Contents............................................................................................................................ ii List of Figures................................................................................................................................. v List of Tables .................................................................................................................................. v List of Attachments........................................................................................................................ vi List of Appendices ......................................................................................................................... vi COMMON UNIT CONVERSIONS AND ABBREVIATIONS................................................... ix CONVERSION OF SCIENTIFIC NOTATION ............................................................................ x EXECUTIVE SUMMARY .......................................................................................................... -
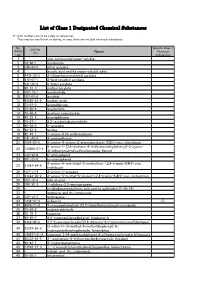
List of Class 1 Designated Chemical Substances
List of Class 1 Designated Chemical Substances *1:CAS numbers are to be solely as references. They may be insufficient or lacking, in case there are multiple chemical substances. No. Specific Class 1 CAS No. (PRTR Chemical (*1) Name Law) Substances 1 - zinc compounds(water-soluble) 2 79-06-1 acrylamide 3 140-88-5 ethyl acrylate 4 - acrylic acid and its water-soluble salts 5 2439-35-2 2-(dimethylamino)ethyl acrylate 6 818-61-1 2-hydroxyethyl acrylate 7 141-32-2 n-butyl acrylate 8 96-33-3 methyl acrylate 9 107-13-1 acrylonitrile 10 107-02-8 acrolein 11 26628-22-8 sodium azide 12 75-07-0 acetaldehyde 13 75-05-8 acetonitrile 14 75-86-5 acetone cyanohydrin 15 83-32-9 acenaphthene 16 78-67-1 2,2'-azobisisobutyronitrile 17 90-04-0 o-anisidine 18 62-53-3 aniline 19 82-45-1 1-amino-9,10-anthraquinone 20 141-43-5 2-aminoethanol 21 1698-60-8 5-amino-4-chloro-2-phenylpyridazin-3(2H)-one; chloridazon 5-amino-1-[2,6-dichloro-4-(trifluoromethyl)phenyl]-3-cyano- 22 120068-37-3 4[(trifluoromethyl)sulfinyl]pyrazole; fipronil 23 123-30-8 p-aminophenol 24 591-27-5 m-aminophenol 4-amino-6-tert-butyl-3-methylthio-1,2,4-triazin-5(4H)-one; 25 21087-64-9 metribuzin 26 107-11-9 3-amino-1-propene 27 41394-05-2 4-amino-3-methyl-6-phenyl-1,2,4-triazin-5(4H)-one; metamitron 28 107-18-6 allyl alcohol 29 106-92-3 1-allyloxy-2,3-epoxypropane 30 - n-alkylbenzenesulfonic acid and its salts(alkyl C=10-14) 31 - antimony and its compounds 32 120-12-7 anthracene 33 1332-21-4 asbestos ○ 34 4098-71-9 3-isocyanatomethyl-3,5,5-trimethylcyclohexyl isocyanate 35 78-84-2 isobutyraldehyde -

Pesticide Resistance of Agricultural Pests in Japan
Pesticide Resistance of Agricultural Pests in Japan By TOSHIKAZU IWATA Chief, 3rd Laboratory of Insect Control Investigation, Department of Phytopathology and Entomology, National Institute of Agricultural Sciences A quarter century has passed since DDT other organophosphorus chemicals such as was firstly applied to control agricultural in methyl paration. As the result the parathion sect pests. The amount of synthesized organic resistant borer was prevented from spreading insecticides applied in fields increased rapidly by changing insecticide to be applied for the since 1952 and the rate of increase was about control. 10 per cent a year. In 1960 an evident report Control of the rice stem borer by BHC dust was made on insecticide resistance of insects, was established in about 1950, and application so 10 years have passed following the first of fine dust or granular formulation of BHC appearance of insecticide-resistant insects. to paddy fields became very popular in 1961 Today, pest species developed or suspected of or 1962. In 1964, declination of susceptibility resistance totals 11 species of insects and 6 of the borers to BHC was found in Kagawa species of mites as shown in Table 1. The Prefecture. After that, similar phenomena author reviews the circumstances of occur were reported in some other prefectures. The rence and some results of studies on major amount of BHC applied to paddy fields has instances of resistant pests. been decreased for its residue in rice plants. Consequently, BHC resistance of rice stem Rice stem borer, Chilo suppressalis borer may not become a serious problem in Walker future. Recently, declination of susceptibility of The rice stem borer is one of the most im the borer to fenitrothion is suspected in a portant pests of rice plants in Japan. -

NMP-Free Formulations of Neonicotinoids
(19) & (11) EP 2 266 400 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 29.12.2010 Bulletin 2010/52 A01N 43/40 (2006.01) A01N 43/86 (2006.01) A01N 47/40 (2006.01) A01N 51/00 (2006.01) (2006.01) (2006.01) (21) Application number: 09305544.0 A01P 7/00 A01N 25/02 (22) Date of filing: 15.06.2009 (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Gasse, Jean-Jacques HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL 27600 Saint-Aubin-Sur-Gaillon (FR) PT RO SE SI SK TR • Duchamp, Guillaume Designated Extension States: 92230 Gennevilliers (FR) AL BA RS • Cantero, Maria 92230 Gennevilliers (FR) (71) Applicant: NUFARM 92233 Gennevelliers (FR) (74) Representative: Cabinet Plasseraud 52, rue de la Victoire 75440 Paris Cedex 09 (FR) (54) NMP-free formulations of neonicotinoids (57) The invention relates to NMP-free liquid formulation comprising at least one nicotinoid and at least one aprotic polar component selected from the group comprising the compounds of formula I, II or III below, and mixtures thereof, wherein R1 and R2 independently represent H or an alkyl group having less than 5 carbons, preferably a methyl group, and n represents an integer ranging from 0 to 5, and to their applications. EP 2 266 400 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 266 400 A1 Description Technical Field of the invention 5 [0001] The invention relates to novel liquid formulations of neonicotinoids and to their use for treating plants, for protecting plants from pests and/or for controlling pests infestation.