Background Information for Chlorpyrifos
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Novel Approach to Fast Determination of 64 Pesticides Using of Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry
Novel approach to fast determination of 64 pesticides using of ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) Tomas Kovalczuk, Ondrej Lacina, Martin Jech, Jan Poustka, Jana Hajslova To cite this version: Tomas Kovalczuk, Ondrej Lacina, Martin Jech, Jan Poustka, Jana Hajslova. Novel approach to fast determination of 64 pesticides using of ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). Food Additives and Contaminants, 2008, 25 (04), pp.444-457. 10.1080/02652030701570156. hal-00577414 HAL Id: hal-00577414 https://hal.archives-ouvertes.fr/hal-00577414 Submitted on 17 Mar 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Food Additives and Contaminants For Peer Review Only Novel approach to fast determination of 64 pesticides using of ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) Journal: Food Additives and Contaminants Manuscript ID: TFAC-2007-065.R1 Manuscript Type: Original Research Paper Date Submitted by the 07-Jul-2007 Author: Complete List of Authors: -

Review of the Mammalian Toxicology
REVIEW OF THE MAMMALIAN TOXICOLOGY AND METABOLISM/TOXICOKINETICS OF FENTHION The APVMA Review of Fenthion © Australian Pesticides and Veterinary Medicines Authority 2012 ISBN 978-0-9873591-5-5 (electronic) This document was originally published in 2005 as part of the preliminary review findings report for Part 1 of the fenthion review. It was subsequently updated in 2007, and revised in 2008. Ownership of intellectual property rights in this publication Unless otherwise noted, copyright (and any other intellectual property rights, if any) in this publication is owned by the Australian Pesticides and Veterinary Medicines Authority (APVMA). Creative Commons licence With the exception of the Coat of Arms, this publication is licensed under a Creative Commons Attribution 3.0 Australia Licence. This is a standard form agreement that allows you to copy, distribute, transmit and adapt this publication provided that you attribute the work. A summary of the licence terms is available from www.creativecommons.org/licenses/by/3.0/au/deed.en. The full licence terms are available from www.creativecommons.org/licenses/by/3.0/au/legalcode. The APVMA’s preference is that you attribute this publication (and any approved material sourced from it) using the following wording: Source: Licensed from the Australian Pesticides and Veterinary Medicines Authority (APVMA) under a Creative Commons Attribution 3.0 Australia Licence. This report was prepared for the APVMA by the Department of Health and Aging Office of Chemical Safety. In referencing this document the Department of Health and Aging Office of Chemical Safety should be cited as the author and the Australian Pesticides and Veterinary Medicines Authority as the publisher and copyright owner. -

Development of a CEN Standardised Method for Liquid Chromatography Coupled to Accurate Mass Spectrometry
Development of a CEN standardised method for liquid chromatography coupled to accurate mass spectrometry CONTENTS 1. Aim and scope ................................................................................................................. 2 2. Short description ................................................................................................................ 2 3. Apparatus and consumables ......................................................................................... 2 4. Chemicals ........................................................................................................................... 2 5. Procedure ........................................................................................................................... 3 5.1. Sample preparation ................................................................................................... 3 5.2. Recovery experiments for method validation ...................................................... 3 5.3. Extraction method ...................................................................................................... 3 5.4. Measurement .............................................................................................................. 3 5.5. Instrumentation and analytical conditions ............................................................ 4 5.5.1. Dionex Ultimate 3000 .......................................................................................... 4 5.5.2. QExactive Focus HESI source parameters ..................................................... -
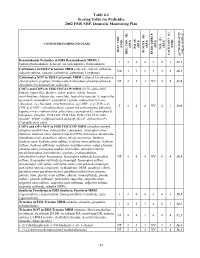
2002 NRP Section 6, Tables 6.1 Through
Table 6.1 Scoring Table for Pesticides 2002 FSIS NRP, Domestic Monitoring Plan } +1 0.05] COMPOUND/COMPOUND CLASS * ) (EPA) (EPA) (EPA) (EPA) (EPA) (FSIS) (FSIS) PSI (P) TOX.(T) L-1 HIST. VIOL. BIOCON. (B) {[( (2*R+P+B)/4]*T} REG. CON. (R) * ENDO. DISRUP. LACK INFO. (L) LACK INFO. {[ Benzimidazole Pesticides in FSIS Benzimidazole MRM (5- 131434312.1 hydroxythiabendazole, benomyl (as carbendazim), thiabendazole) Carbamates in FSIS Carbamate MRM (aldicarb, aldicarb sulfoxide, NA44234416.1 aldicarb sulfone, carbaryl, carbofuran, carbofuran 3-hydroxy) Carbamates NOT in FSIS Carbamate MRM (carbaryl 5,6-dihydroxy, chlorpropham, propham, thiobencarb, 4-chlorobenzylmethylsulfone,4- NT 4 1 3 NV 4 4 13.8 chlorobenzylmethylsulfone sulfoxide) CHC's and COP's in FSIS CHC/COP MRM (HCB, alpha-BHC, lindane, heptachlor, dieldrin, aldrin, endrin, ronnel, linuron, oxychlordane, chlorpyrifos, nonachlor, heptachlor epoxide A, heptachlor epoxide B, endosulfan I, endosulfan I sulfate, endosulfan II, trans- chlordane, cis-chlordane, chlorfenvinphos, p,p'-DDE, p, p'-TDE, o,p'- 3444NV4116.0 DDT, p,p'-DDT, carbophenothion, captan, tetrachlorvinphos [stirofos], kepone, mirex, methoxychlor, phosalone, coumaphos-O, coumaphos-S, toxaphene, famphur, PCB 1242, PCB 1248, PCB 1254, PCB 1260, dicofol*, PBBs*, polybrominated diphenyl ethers*, deltamethrin*) (*identification only) COP's and OP's NOT in FSIS CHC/COP MRM (azinphos-methyl, azinphos-methyl oxon, chlorpyrifos, coumaphos, coumaphos oxon, diazinon, diazinon oxon, diazinon met G-27550, dichlorvos, dimethoate, dimethoate -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Assessment of the Endocrine-Disrupting Effects Of
Food and Chemical Toxicology 133 (2019) 110759 Contents lists available at ScienceDirect Food and Chemical Toxicology journal homepage: www.elsevier.com/locate/foodchemtox Assessment of the endocrine-disrupting effects of organophosphorus T pesticide triazophos and its metabolites on endocrine hormones biosynthesis, transport and receptor binding in silico ⁎ Fang-Wei Yanga, Yi-Xuan Lia, Fa-Zheng Rena,b, Jie Luoa,c, Guo-Fang Panga,d, a Beijing Advanced Innovation Center for Food Nutrition and Human Health, College of Food Science and Nutritional Engineering, China Agricultural University, Beijing, 100083, China b Key Laboratory of Functional Dairy, Co-constructed by Ministry of Education and Beijing Government, Beijing Laboratory of Food Quality and Safety, China Agricultural University, Beijing, 100083, China c College of Food Science and Technology, Hunan Agricultural University, Changsha, 410114, China d Chinese Academy of Inspection and Quarantine, Beijing, 100176, China ARTICLE INFO ABSTRACT Keywords: Triazophos (TAP) was a widely used organophosphorus insecticide in developing countries. TAP could produce Triazophos specific metabolites triazophos-oxon (TAPO) and 1-phenyl-3-hydroxy-1,2,4-triazole (PHT) and non-specific Metabolites metabolites diethylthiophosphate (DETP) and diethylphosphate (DEP). The objective of this study involved Endocrine disruption computational approaches to discover potential mechanisms of molecular interaction of TAP and its major Hormones metabolites with endocrine hormone-related proteins using molecular docking in silico. We found that TAP, Molecular docking TAPO and DEP showed high binding affinity with more proteins and enzymes than PHT and DETP. TAPmight interfere with the endocrine function of the adrenal gland, and TAP might also bind strongly with glucocorticoid receptors and thyroid hormone receptors. -

Eupt-Fv17- Target Pesticide List
EUPT-FV17- TARGET PESTICIDE LIST MRRL Pesticide (mg/Kg) 3-hydroxy-carbofuran 0.01 Acephate 0.01 Acetamiprid 0.01 Acrinathrin 0.01 Aldicarb 0.01 Aldicarb Sulfone 0.01 Aldicarb Sulfoxide 0.01 Azinphos-methyl 0.01 Azoxystrobin 0.01 Benfuracarb 0.01 Benomyl 0.01 Bifenthrin 0.01 Bitertanol 0.01 Boscalid 0.01 Bromopropylate 0.01 Bromuconazole 0.01 Bupirimate 0.01 Buprofezin 0.01 Cadusafos 0.006 Carbaryl 0.01 Carbendazim 0.01 Carbofuran 0.01 Carbosulfan 0.01 Chlorfenapyr 0.01 Chlorfenvinphos 0.01 Chlorobenzilate 0.01 Chlorothalonil 0.01 Chlorpropham (only parent compound) 0.01 Chlorpyrifos 0.01 Chlorpyrifos-methyl 0.01 Clofentezine (only parent compound) 0.01 Clothianidin 0.01 Cyfluthrin (cyfluthrin incl. other mixtures of constituent isomers (sum of isomers)) 0.01 Cypermethrin (cypermethrin incl. other mixtures of constituent isomers (sum of isomers)) 0.01 Cyproconazole 0.01 Cyprodinil 0.01 Deltamethrin 0.01 Demeton-S-methylsulfone 0.006 Desmethyl-pirimicarb 0.01 Diazinon 0.01 Dichlofluanid (only parent compound) 0.01 Dichlorvos 0.01 Dicloran 0.01 Dicofol 0.01 Diethofencarb 0.01 Difenoconazole 0.01 Diflubenzuron 0.01 Dimethoate 0.003 Dimethomorph 0.01 Dimethylaminosulfotoluidide (DMST) 0.01 Diniconazole 0.01 Diphenylamine 0.01 Endosulfan alpha 0.01 Endosulfan beta 0.01 Endosulfan sulfate 0.01 EPN 0.01 Epoxiconazole 0.01 Ethion 0.01 Ethirimol 0.01 Ethoprophos 0.008 Etofenprox 0.01 Fenamidone 0.01 Fenamiphos 0.01 Fenamiphos sulfone 0.01 Fenamiphos sulfoxide 0.01 Fenarimol 0.01 Fenazaquin 0.01 Fenbuconazole 0.01 Fenhexamid 0.01 Fenitrothion 0.01 -

744 Hydrolysis of Chiral Organophosphorus Compounds By
[Frontiers in Bioscience, Landmark, 26, 744-770, Jan 1, 2021] Hydrolysis of chiral organophosphorus compounds by phosphotriesterases and mammalian paraoxonase-1 Antonio Monroy-Noyola1, Damianys Almenares-Lopez2, Eugenio Vilanova Gisbert3 1Laboratorio de Neuroproteccion, Facultad de Farmacia, Universidad Autonoma del Estado de Morelos, Morelos, Mexico, 2Division de Ciencias Basicas e Ingenierias, Universidad Popular de la Chontalpa, H. Cardenas, Tabasco, Mexico, 3Instituto de Bioingenieria, Universidad Miguel Hernandez, Elche, Alicante, Spain TABLE OF CONTENTS 1. Abstract 2. Introduction 2.1. Organophosphorus compounds (OPs) and their toxicity 2.2. Metabolism and treatment of OP intoxication 2.3. Chiral OPs 3. Stereoselective hydrolysis 3.1. Stereoselective hydrolysis determines the toxicity of chiral compounds 3.2. Hydrolysis of nerve agents by PTEs 3.2.1. Hydrolysis of V-type agents 3.3. PON1, a protein restricted in its ability to hydrolyze chiral OPs 3.4. Toxicity and stereoselective hydrolysis of OPs in animal tissues 3.4.1. The calcium-dependent stereoselective activity of OPs associated with PON1 3.4.2. Stereoselective hydrolysis commercial OPs pesticides by alloforms of PON1 Q192R 3.4.3. PON1, an enzyme that stereoselectively hydrolyzes OP nerve agents 3.4.4. PON1 recombinants and stereoselective hydrolysis of OP nerve agents 3.5. The activity of PTEs in birds 4. Conclusions 5. Acknowledgments 6. References 1. ABSTRACT Some organophosphorus compounds interaction of the racemic OPs with these B- (OPs), which are used in the manufacturing of esterases (AChE and NTE) and such interactions insecticides and nerve agents, are racemic mixtures have been studied in vivo, ex vivo and in vitro, using with at least one chiral center with a phosphorus stereoselective hydrolysis by A-esterases or atom. -

Organophosphate Insecticides
CHAPTER 4 HIGHLIGHTS Organophosphate Insecticides Acts through phosphorylation of the acetylcholinesterase enzyme Since the removal of organochlorine insecticides from use, organophosphate at nerve endings insecticides have become the most widely used insecticides available today. More Absorbed by inhalation, than forty of them are currently registered for use and all run the risk of acute ingestion, and skin and subacute toxicity. Organophosphates are used in agriculture, in the home, penetration in gardens, and in veterinary practice. All apparently share a common mecha- Muscarinic, nicotinic & CNS nism of cholinesterase inhibition and can cause similar symptoms. Because they effects share this mechanism, exposure to the same organophosphate by multiple routes or to multiple organophosphates by multiple routes can lead to serious additive Signs and Symptoms: toxicity. It is important to understand, however, that there is a wide range of Headache, hypersecretion, toxicity in these agents and wide variation in cutaneous absorption, making muscle twitching, nausea, specific identification and management quite important. diarrhea Respiratory depression, seizures, loss of consciousness Toxicology Miosis is often a helpful Organophosphates poison insects and mammals primarily by phosphory- diagnostic sign lation of the acetylcholinesterase enzyme (AChE) at nerve endings. The result is a loss of available AChE so that the effector organ becomes overstimulated by Treatment: the excess acetylcholine (ACh, the impulse-transmitting substance) in the nerve Clear airway, improve tissue ending. The enzyme is critical to normal control of nerve impulse transmission oxygenation from nerve fibers to smooth and skeletal muscle cells, glandular cells, and Administer atropine sulfate autonomic ganglia, as well as within the central nervous system (CNS). -

Acutely / Extremely Hazardous Waste List
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extemely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extemely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extemely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extemely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extemely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extemely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extemely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extemely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extemely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Qsar Analysis of the Chemical Hydrolysis of Organophosphorus Pesticides in Natural Waters
QSAR ANALYSIS OF THE CHEMICAL HYDROLYSIS OF ORGANOPHOSPHORUS PESTICIDES IN NATURAL WATERS. by Kenneth K. Tanji Principal Investigator and Jonathan 1. Sullivan Graduate Research Assistant Department of Land, Air and Water Resources University of California, Davis Technical Completion Report Project Number W-843 August, 1995 University of California Water Resource Center The research leading to this report was supported by the University of California Water Resource Center as part of Water Resource Center Project W-843. Table of Contents Page Abstract 2 Problem and Research Objectives 3 Introduction 5 Theoretical Background 6 QSAR Methodology 7 Molecular Connectivity Theory 8 Organophosphorus Pesticides 12 Experimental Determination of Rates 15 Results and Discussion 17 Principal Findings and Significance 19 References 34 List of Tables Page Table 1. Statistical relationship between OP pesticides and first-order MC/'s. 30 Table 2. Inherent conditions of waters used in experimental work. 16 Table 3. Estimated half-lives for organophosphorus esters derived from model. 31 Table 4. Half-lives and first-order MCI' sfor model calibration data set. 31 Table 5. Experimental kinetic data for validation set compounds, Sacramento. 33 List of Figures Page Figure 1. Essential Features OfQSAR Modeling Methodology. 21 Figure 2. Regression plot for In hydrolysis rate vs. 1st order MCl' s. 22 Figure 3. a 3-D molecular model, a line-segment model and a graphical model. 23 Figure 4. Molecular connectivity index suborders. 24 Figure 5. Chlorpyrifos and its fourteen fourth order path/cluster fragments. 25 Figure 6. Abridged MClndex output. 26 Figure 7. Parent acids of most common organophosphorus pesticides. 12 Figure 8. -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination