Novel Approach to Fast Determination of 64 Pesticides Using of Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Development of a CEN Standardised Method for Liquid Chromatography Coupled to Accurate Mass Spectrometry
Development of a CEN standardised method for liquid chromatography coupled to accurate mass spectrometry CONTENTS 1. Aim and scope ................................................................................................................. 2 2. Short description ................................................................................................................ 2 3. Apparatus and consumables ......................................................................................... 2 4. Chemicals ........................................................................................................................... 2 5. Procedure ........................................................................................................................... 3 5.1. Sample preparation ................................................................................................... 3 5.2. Recovery experiments for method validation ...................................................... 3 5.3. Extraction method ...................................................................................................... 3 5.4. Measurement .............................................................................................................. 3 5.5. Instrumentation and analytical conditions ............................................................ 4 5.5.1. Dionex Ultimate 3000 .......................................................................................... 4 5.5.2. QExactive Focus HESI source parameters ..................................................... -
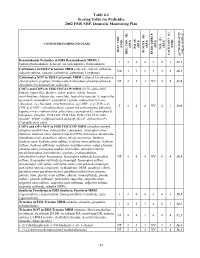
2002 NRP Section 6, Tables 6.1 Through
Table 6.1 Scoring Table for Pesticides 2002 FSIS NRP, Domestic Monitoring Plan } +1 0.05] COMPOUND/COMPOUND CLASS * ) (EPA) (EPA) (EPA) (EPA) (EPA) (FSIS) (FSIS) PSI (P) TOX.(T) L-1 HIST. VIOL. BIOCON. (B) {[( (2*R+P+B)/4]*T} REG. CON. (R) * ENDO. DISRUP. LACK INFO. (L) LACK INFO. {[ Benzimidazole Pesticides in FSIS Benzimidazole MRM (5- 131434312.1 hydroxythiabendazole, benomyl (as carbendazim), thiabendazole) Carbamates in FSIS Carbamate MRM (aldicarb, aldicarb sulfoxide, NA44234416.1 aldicarb sulfone, carbaryl, carbofuran, carbofuran 3-hydroxy) Carbamates NOT in FSIS Carbamate MRM (carbaryl 5,6-dihydroxy, chlorpropham, propham, thiobencarb, 4-chlorobenzylmethylsulfone,4- NT 4 1 3 NV 4 4 13.8 chlorobenzylmethylsulfone sulfoxide) CHC's and COP's in FSIS CHC/COP MRM (HCB, alpha-BHC, lindane, heptachlor, dieldrin, aldrin, endrin, ronnel, linuron, oxychlordane, chlorpyrifos, nonachlor, heptachlor epoxide A, heptachlor epoxide B, endosulfan I, endosulfan I sulfate, endosulfan II, trans- chlordane, cis-chlordane, chlorfenvinphos, p,p'-DDE, p, p'-TDE, o,p'- 3444NV4116.0 DDT, p,p'-DDT, carbophenothion, captan, tetrachlorvinphos [stirofos], kepone, mirex, methoxychlor, phosalone, coumaphos-O, coumaphos-S, toxaphene, famphur, PCB 1242, PCB 1248, PCB 1254, PCB 1260, dicofol*, PBBs*, polybrominated diphenyl ethers*, deltamethrin*) (*identification only) COP's and OP's NOT in FSIS CHC/COP MRM (azinphos-methyl, azinphos-methyl oxon, chlorpyrifos, coumaphos, coumaphos oxon, diazinon, diazinon oxon, diazinon met G-27550, dichlorvos, dimethoate, dimethoate -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Assessment of the Endocrine-Disrupting Effects Of
Food and Chemical Toxicology 133 (2019) 110759 Contents lists available at ScienceDirect Food and Chemical Toxicology journal homepage: www.elsevier.com/locate/foodchemtox Assessment of the endocrine-disrupting effects of organophosphorus T pesticide triazophos and its metabolites on endocrine hormones biosynthesis, transport and receptor binding in silico ⁎ Fang-Wei Yanga, Yi-Xuan Lia, Fa-Zheng Rena,b, Jie Luoa,c, Guo-Fang Panga,d, a Beijing Advanced Innovation Center for Food Nutrition and Human Health, College of Food Science and Nutritional Engineering, China Agricultural University, Beijing, 100083, China b Key Laboratory of Functional Dairy, Co-constructed by Ministry of Education and Beijing Government, Beijing Laboratory of Food Quality and Safety, China Agricultural University, Beijing, 100083, China c College of Food Science and Technology, Hunan Agricultural University, Changsha, 410114, China d Chinese Academy of Inspection and Quarantine, Beijing, 100176, China ARTICLE INFO ABSTRACT Keywords: Triazophos (TAP) was a widely used organophosphorus insecticide in developing countries. TAP could produce Triazophos specific metabolites triazophos-oxon (TAPO) and 1-phenyl-3-hydroxy-1,2,4-triazole (PHT) and non-specific Metabolites metabolites diethylthiophosphate (DETP) and diethylphosphate (DEP). The objective of this study involved Endocrine disruption computational approaches to discover potential mechanisms of molecular interaction of TAP and its major Hormones metabolites with endocrine hormone-related proteins using molecular docking in silico. We found that TAP, Molecular docking TAPO and DEP showed high binding affinity with more proteins and enzymes than PHT and DETP. TAPmight interfere with the endocrine function of the adrenal gland, and TAP might also bind strongly with glucocorticoid receptors and thyroid hormone receptors. -

Eupt-Fv17- Target Pesticide List
EUPT-FV17- TARGET PESTICIDE LIST MRRL Pesticide (mg/Kg) 3-hydroxy-carbofuran 0.01 Acephate 0.01 Acetamiprid 0.01 Acrinathrin 0.01 Aldicarb 0.01 Aldicarb Sulfone 0.01 Aldicarb Sulfoxide 0.01 Azinphos-methyl 0.01 Azoxystrobin 0.01 Benfuracarb 0.01 Benomyl 0.01 Bifenthrin 0.01 Bitertanol 0.01 Boscalid 0.01 Bromopropylate 0.01 Bromuconazole 0.01 Bupirimate 0.01 Buprofezin 0.01 Cadusafos 0.006 Carbaryl 0.01 Carbendazim 0.01 Carbofuran 0.01 Carbosulfan 0.01 Chlorfenapyr 0.01 Chlorfenvinphos 0.01 Chlorobenzilate 0.01 Chlorothalonil 0.01 Chlorpropham (only parent compound) 0.01 Chlorpyrifos 0.01 Chlorpyrifos-methyl 0.01 Clofentezine (only parent compound) 0.01 Clothianidin 0.01 Cyfluthrin (cyfluthrin incl. other mixtures of constituent isomers (sum of isomers)) 0.01 Cypermethrin (cypermethrin incl. other mixtures of constituent isomers (sum of isomers)) 0.01 Cyproconazole 0.01 Cyprodinil 0.01 Deltamethrin 0.01 Demeton-S-methylsulfone 0.006 Desmethyl-pirimicarb 0.01 Diazinon 0.01 Dichlofluanid (only parent compound) 0.01 Dichlorvos 0.01 Dicloran 0.01 Dicofol 0.01 Diethofencarb 0.01 Difenoconazole 0.01 Diflubenzuron 0.01 Dimethoate 0.003 Dimethomorph 0.01 Dimethylaminosulfotoluidide (DMST) 0.01 Diniconazole 0.01 Diphenylamine 0.01 Endosulfan alpha 0.01 Endosulfan beta 0.01 Endosulfan sulfate 0.01 EPN 0.01 Epoxiconazole 0.01 Ethion 0.01 Ethirimol 0.01 Ethoprophos 0.008 Etofenprox 0.01 Fenamidone 0.01 Fenamiphos 0.01 Fenamiphos sulfone 0.01 Fenamiphos sulfoxide 0.01 Fenarimol 0.01 Fenazaquin 0.01 Fenbuconazole 0.01 Fenhexamid 0.01 Fenitrothion 0.01 -

744 Hydrolysis of Chiral Organophosphorus Compounds By
[Frontiers in Bioscience, Landmark, 26, 744-770, Jan 1, 2021] Hydrolysis of chiral organophosphorus compounds by phosphotriesterases and mammalian paraoxonase-1 Antonio Monroy-Noyola1, Damianys Almenares-Lopez2, Eugenio Vilanova Gisbert3 1Laboratorio de Neuroproteccion, Facultad de Farmacia, Universidad Autonoma del Estado de Morelos, Morelos, Mexico, 2Division de Ciencias Basicas e Ingenierias, Universidad Popular de la Chontalpa, H. Cardenas, Tabasco, Mexico, 3Instituto de Bioingenieria, Universidad Miguel Hernandez, Elche, Alicante, Spain TABLE OF CONTENTS 1. Abstract 2. Introduction 2.1. Organophosphorus compounds (OPs) and their toxicity 2.2. Metabolism and treatment of OP intoxication 2.3. Chiral OPs 3. Stereoselective hydrolysis 3.1. Stereoselective hydrolysis determines the toxicity of chiral compounds 3.2. Hydrolysis of nerve agents by PTEs 3.2.1. Hydrolysis of V-type agents 3.3. PON1, a protein restricted in its ability to hydrolyze chiral OPs 3.4. Toxicity and stereoselective hydrolysis of OPs in animal tissues 3.4.1. The calcium-dependent stereoselective activity of OPs associated with PON1 3.4.2. Stereoselective hydrolysis commercial OPs pesticides by alloforms of PON1 Q192R 3.4.3. PON1, an enzyme that stereoselectively hydrolyzes OP nerve agents 3.4.4. PON1 recombinants and stereoselective hydrolysis of OP nerve agents 3.5. The activity of PTEs in birds 4. Conclusions 5. Acknowledgments 6. References 1. ABSTRACT Some organophosphorus compounds interaction of the racemic OPs with these B- (OPs), which are used in the manufacturing of esterases (AChE and NTE) and such interactions insecticides and nerve agents, are racemic mixtures have been studied in vivo, ex vivo and in vitro, using with at least one chiral center with a phosphorus stereoselective hydrolysis by A-esterases or atom. -

Organophosphate Insecticides
CHAPTER 4 HIGHLIGHTS Organophosphate Insecticides Acts through phosphorylation of the acetylcholinesterase enzyme Since the removal of organochlorine insecticides from use, organophosphate at nerve endings insecticides have become the most widely used insecticides available today. More Absorbed by inhalation, than forty of them are currently registered for use and all run the risk of acute ingestion, and skin and subacute toxicity. Organophosphates are used in agriculture, in the home, penetration in gardens, and in veterinary practice. All apparently share a common mecha- Muscarinic, nicotinic & CNS nism of cholinesterase inhibition and can cause similar symptoms. Because they effects share this mechanism, exposure to the same organophosphate by multiple routes or to multiple organophosphates by multiple routes can lead to serious additive Signs and Symptoms: toxicity. It is important to understand, however, that there is a wide range of Headache, hypersecretion, toxicity in these agents and wide variation in cutaneous absorption, making muscle twitching, nausea, specific identification and management quite important. diarrhea Respiratory depression, seizures, loss of consciousness Toxicology Miosis is often a helpful Organophosphates poison insects and mammals primarily by phosphory- diagnostic sign lation of the acetylcholinesterase enzyme (AChE) at nerve endings. The result is a loss of available AChE so that the effector organ becomes overstimulated by Treatment: the excess acetylcholine (ACh, the impulse-transmitting substance) in the nerve Clear airway, improve tissue ending. The enzyme is critical to normal control of nerve impulse transmission oxygenation from nerve fibers to smooth and skeletal muscle cells, glandular cells, and Administer atropine sulfate autonomic ganglia, as well as within the central nervous system (CNS). -

Background Information for Chlorpyrifos
64 Appendix A: Background Information for Chlorpyrifos Chlorpyrifos is an organophosphorus insecticide. It belongs to the phosphorothioate (also called phosphorothionate) group, composed of organophosphorus compounds that contain the P=S substructure. Other organophosphorus insecticides in this group include diazinon, methyl parathion, parathion, fenthion, and fenitrothion. These compounds require metabolic activation to their oxon analogs (compounds in which the =S is replaced by =O) for anticholinesterase activity. The structures of chlorpyrifos and its toxic metabolite, chlorpyrifos oxon, are provided in Appendix D. As of 2000, chlorpyrifos was one of the most widely used organophosphorus insecticides in the United States, for both agricultural and residential purposes. Registered uses included food crops, turf and ornamental plants, indoor pest control (including crack and crevice treatment), termite control, mosquito control, and pet collars. It was registered for use in a wide variety of buildings, including residential, commercial, schools, daycare centers, restaurants, hospitals, hotels, and food manufacturing plants (EPA 2000c). Many uses of chlorpyrifos are being phased out (see Section A.4). A study of urinary pesticide metabolites during the third trimester in 386 pregnant women from East Harlem indicated that exposure to chlorpyrifos was prevalent (42% of the women had detectable levels of the chlorpyrifos metabolite), was higher than the median in NHANES III, did not show seasonal variation, and did not change during the -

Acutely / Extremely Hazardous Waste List
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extemely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extemely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extemely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extemely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extemely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extemely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extemely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extemely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extemely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination -

The Importance of Metabolites in Trace Organic Residue Analysis
the importance of metabolites in trace organic residue analysis lgcstandards.com/drehrenstorfer [email protected] LGC Quality | ISO 17034 | ISO/IEC 17025 | ISO 9001 INTRODUCTION Many organic contaminants of concern in the environment and food chain are biologically active. This includes residues of pesticides, veterinary medicines and human pharmaceuticals. Almost by definition, biologically active chemicals are subject There will be a range of metabolites and degradation products, to metabolism by animals, plants or microorganisms. which may or may not be of significance. In the case of Metabolites are formed as a by-product of the biological registered active substances with legal Maximum Residue effect, or as a mechanism for an organism to absorb or Levels (MRLs) in food - i.e. pesticides and veterinary medicines excrete the chemical. This mechanism may be obvious - an assessment will have been made if any metabolites are in the case of animals or plants, or less so in the case of significant in terms of either relative concentration or environmental degradation by the action of microorganisms. toxicology, and the MRL will define which metabolites (if any) Metabolites and degradation products are often much more must be measured and summed. This paper uses a series persistent than the parent chemical. Residues of biologically of examples to illustrate the significance of metabolites in active chemicals in food or in the environment rarely exist interpreting analytical results, and different approaches to purely as the parent compound. their analytical measurement. 2 lgcstandards.com/drehrenstorfer SHOULD ALL METABOLITES BE ANALYSED? Metabolites are not always significant, and there is often no need to analyse them. -

Spider Mites and Resistance 653 in Production and Better Flowers V/Ith Quired Dosages
day to nearly 2 days. Then it enters the resting stage. After a day or so it molts to a second active stage, which feeds Spider Mites and and then becomes quiescent as in the first stage. The adult male emerges Resistance from the second quiescent stage, but the female passes through a third feed- Floyd F. Smith ing and quiescent stage before becom- ing an adult. Mating usually occurs within a few minutes after the female Spider mites, or "red spiders," at- becomes an adult. Only males develop tack nearly all kinds of field crops, veg- from eggs of unmated females. etables, orchard trees, weeds, and The developmental period varies greenhouse plants. The mites often v-^-idely with the temperature. The eggs confine themselves to one kind of plant hatch in 2 or 3 days at 75° F. or higher at first but move to other kinds when or after 21 days at 55°. The mites may injury increases and food becomes reach the adult stage within 5 days at scarce. 75° or in 40 days at 55°. Under aver- Several species, of similar habits but age greenhouse temperatures of 60° to different morphological characters, arc 70°, the incubation period is 5 to 10 found out of doors. days and development to adult stage Of six species that infest green- from 10 to 15 days. One female lays a houses, the two-spotted spider mite is few eggs daily and a total of 100 to predominant. It is a general feeder 194 eggs during an average life of 3 to but it is almost constantly present on 4 weeks.