Accurate and Efficient Detection of Gene Fusions from RNA Sequencing Data
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Evolution and Structure of Clinically Relevant Gene Fusions in Multiple Myeloma
ARTICLE https://doi.org/10.1038/s41467-020-16434-y OPEN Evolution and structure of clinically relevant gene fusions in multiple myeloma Steven M. Foltz 1,2, Qingsong Gao 1,2, Christopher J. Yoon1,2, Hua Sun1,2, Lijun Yao1,2, Yize Li1,2, ✉ ✉ Reyka G. Jayasinghe1,2, Song Cao1,2, Justin King1, Daniel R. Kohnen1, Mark A. Fiala1, Li Ding1,2,3,4 & Ravi Vij1,4 Multiple myeloma is a plasma cell blood cancer with frequent chromosomal translocations leading to gene fusions. To determine the clinical relevance of fusion events, we detect gene 1234567890():,; fusions from a cohort of 742 patients from the Multiple Myeloma Research Foundation CoMMpass Study. Patients with multiple clinic visits enable us to track tumor and fusion evolution, and cases with matching peripheral blood and bone marrow samples allow us to evaluate the concordance of fusion calls in patients with high tumor burden. We examine the joint upregulation of WHSC1 and FGFR3 in samples with t(4;14)-related fusions, and we illustrate a method for detecting fusions from single cell RNA-seq. We report fusions at MYC and a neighboring gene, PVT1, which are related to MYC translocations and associated with divergent progression-free survival patterns. Finally, we find that 4% of patients may be eligible for targeted fusion therapies, including three with an NTRK1 fusion. 1 Department of Medicine, Washington University in St. Louis, St. Louis, MO 63110, USA. 2 McDonnell Genome Institute, Washington University in St. Louis, St. Louis, MO 63108, USA. 3 Department of Genetics, Washington University in St. Louis, St. -

Tumour-Agnostic Therapy for Pancreatic Cancer and Biliary Tract Cancer
diagnostics Review Tumour-Agnostic Therapy for Pancreatic Cancer and Biliary Tract Cancer Shunsuke Kato Department of Clinical Oncology, Juntendo University Graduate School of Medicine, 2-1-1, Hongo, Bunkyo-ku, Tokyo 113-8421, Japan; [email protected]; Tel.: +81-3-5802-1543 Abstract: The prognosis of patients with solid tumours has remarkably improved with the develop- ment of molecular-targeted drugs and immune checkpoint inhibitors. However, the improvements in the prognosis of pancreatic cancer and biliary tract cancer is delayed compared to other carcinomas, and the 5-year survival rates of distal-stage disease are approximately 10 and 20%, respectively. How- ever, a comprehensive analysis of tumour cells using The Cancer Genome Atlas (TCGA) project has led to the identification of various driver mutations. Evidently, few mutations exist across organs, and basket trials targeting driver mutations regardless of the primary organ are being actively conducted. Such basket trials not only focus on the gate keeper-type oncogene mutations, such as HER2 and BRAF, but also focus on the caretaker-type tumour suppressor genes, such as BRCA1/2, mismatch repair-related genes, which cause hereditary cancer syndrome. As oncogene panel testing is a vital approach in routine practice, clinicians should devise a strategy for improved understanding of the cancer genome. Here, the gene mutation profiles of pancreatic cancer and biliary tract cancer have been outlined and the current status of tumour-agnostic therapy in these cancers has been reported. Keywords: pancreatic cancer; biliary tract cancer; targeted therapy; solid tumours; driver mutations; agonist therapy Citation: Kato, S. Tumour-Agnostic Therapy for Pancreatic Cancer and 1. -

Procedure of Protoplast Electrofusion (In Solanum Genus)
Procedure of protoplast electrofusion (in Solanum genus) A part of the output V002 within the project NAZV QF4108 “Use of protoplast fusion technique in breeding of economic important crops of Brassica, Cucumis and Solanum genus” applied and verified in the solution of MSM 6010980701 “Molecular and technological basis of quality potato production” Authors: Ing. Marie Greplová Mgr. Hana Polzerová Ing. Jaroslava Domkářová, Ph.D., MBA 1 Introduction The effective development of new varieties with desirable traits requires a combination of conventional breeding approaches and modern technologies such as in vitro cultures, involving production of dihaploid lines, protoplast fusions and use of genetic transformation. Modern plant biotechnologies are a necessary part of plant breeding for resistance to biotic and abiotic stresses. They contribute to an enlargement of variety spectrum and to decrease inputs for profitable yields. Breeding (sexual hybridization) of economic important crops for resistance to biotic and abiotic agents is often interfered with many complications as sexual incompatibility of parental genotypes, unsuitable EBN ratio (Endosperm Balance Number, Carputo et al. 1997) or small success in conventional sexual hybridization. Problems with resistance transfer in classical approaches resulted in development of unconventional approaches. Interspecific or intergeneric protoplast fusions offer an alternative way of gene transfer (Millam et al. 1995). Since there are no barriers to protoplast fusion, hitherto incompatible and therefore reproductively isolated species, can be brought together at the protoplast level. Following fusion, heterocaryons, containing nuclei of two species in common cytoplasm regenerate a new cell wall, enter division and nuclear fusion results in the formation of a somatic hybrid cell. -

Protoplast Cultures and Protoplast Fusion Focused on Brassicaceae – a Review
Protoplast cultures and protoplast fusion focused on Brassicaceae – a review B. NAVRÁTILOVÁ Faculty of Science, Palacký University in Olomouc, Olomouc, Czech Republic ABSTRACT: The subjects of this article are protoplast isolations and protoplast fusions, in particular their history, a review of factors influencing the protoplasts isolation and fusion, selection of hybrid plants and utilization of somatic hybrids in plant breed- ing. Somatic hybridization through protoplast fusion can overcome sexual incompatibility among plant species or genera; transfer genes of resistance to diseases (viral, bacterial, fungal), pests, herbicides and others stress factors; obtain cybrid plants; transfer cytoplasmic male sterility or incease content of secondary metabolites in hybrid plants. The article is focussed mainly on the family Brassicaceae because among representatives are significant crops for the human population. Various successful combination of intraspecific, interspecific and intergeneric protoplast fusion were reported between representatives of the family Brassicaceae with the genus Brassica which belonged to the first agricultural crops used for the isolation of protoplast. Keywords: Brassicaceae; Brassica; protoplast isolation; protoplast fusion; somatic hybridization; resistance to diseases and abiotic factors Isolated protoplasts are a unique system for studying interspecific hybrid obtained by the protoplast fusion of the structure and function of cell organelles, cytoplasmic Nicotiana glauca + Nicotiana langsdorffii appeared in membrane transport in plants and cell wall formation. 1972 (CARLSON et al. 1972), but this hybrid could also Another possibility of their use is protoplast fusions, ge- be produced by sexual crossing. The finding was that the netic manipulations or experimental mutagenesis. hybrids were characterized by spontaneous emergence Somatic hybridization through plant protoplast fu- of tumours without any need to add growth regulators sion enables not only to combine parent genes in higher into the culture medium. -

DNAJB1–PRKACA Fusion Kinase Interacts with Β-Catenin and the Liver
DNAJB1–PRKACA fusion kinase interacts with INAUGURAL ARTICLE β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma Edward R. Kastenhubera,b, Gadi Lalazarc, Shauna L. Houlihana, Darjus F. Tschaharganehd,e, Timour Baslana, Chi-Chao Chena, David Requenac, Sha Tiana, Benedikt Bosbachf, John E. Wilkinsong, Sanford M. Simonc, and Scott W. Lowea,h,1 aDepartment of Cancer Biology and Genetics, Memorial Sloan Kettering Cancer Center, New York, NY 10065; bLouis V. Gerstner Jr. Graduate School of Biomedical Sciences, Memorial Sloan Kettering Cancer Center, New York, NY 10065; cLaboratory of Cellular Biophysics, The Rockefeller University, New York, NY 10065; dHelmholtz University Group “Cell Plasticity and Epigenetic Remodeling,” German Cancer Research Center (DKFZ), 69120 Heidelberg, Germany; eInstitute of Pathology, University Hospital, 69120 Heidelberg, Germany; fOncology Target Discovery Program, Pfizer Inc., Pearl River, NY 10965; gDepartment of Pathology, University of Michigan School of Medicine, Ann Arbor, MI 48109; and hHoward Hughes Medical Institute, New York, NY 10065 This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected in 2017. Contributed by Scott W. Lowe, October 26, 2017 (sent for review September 22, 2017; reviewed by Nabeel M. Bardeesy and David A. Largaespada) A segmental deletion resulting in DNAJB1–PRKACA gene fusion is any known etiological risk factors such as alcoholism, chronic hep- now recognized as the signature genetic event of fibrolamellar hepa- atitis infection, or liver flukes (8). tocellular carcinoma (FL-HCC), a rare but lethal liver cancer that pri- Currently, FL-HCC is diagnosed on the basis of histological marily affects adolescents and young adults. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

PRKACA Mediates Resistance to HER2-Targeted Therapy in Breast Cancer Cells and Restores Anti-Apoptotic Signaling
Oncogene (2015) 34, 2061–2071 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc ORIGINAL ARTICLE PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling SE Moody1,2,3, AC Schinzel1, S Singh1, F Izzo1, MR Strickland1, L Luo1,2, SR Thomas3, JS Boehm3, SY Kim4, ZC Wang5,6 and WC Hahn1,2,3 Targeting HER2 with antibodies or small molecule inhibitors in HER2-positive breast cancer leads to improved survival, but resistance is a common clinical problem. To uncover novel mechanisms of resistance to anti-HER2 therapy in breast cancer, we performed a kinase open reading frame screen to identify genes that rescue HER2-amplified breast cancer cells from HER2 inhibition or suppression. In addition to multiple members of the MAPK (mitogen-activated protein kinase) and PI3K (phosphoinositide 3-kinase) signaling pathways, we discovered that expression of the survival kinases PRKACA and PIM1 rescued cells from anti-HER2 therapy. Furthermore, we observed elevated PRKACA expression in trastuzumab-resistant breast cancer samples, indicating that this pathway is activated in breast cancers that are clinically resistant to trastuzumab-containing therapy. We found that neither PRKACA nor PIM1 restored MAPK or PI3K activation after lapatinib or trastuzumab treatment, but rather inactivated the pro-apoptotic protein BAD, the BCl-2-associated death promoter, thereby permitting survival signaling through BCL- XL. Pharmacological blockade of BCL-XL/BCL-2 partially abrogated the rescue effects conferred by PRKACA and PIM1, and sensitized cells to lapatinib treatment. These observations suggest that combined targeting of HER2 and the BCL-XL/BCL-2 anti-apoptotic pathway may increase responses to anti-HER2 therapy in breast cancer and decrease the emergence of resistant disease. -

A Novel Resveratrol Analog: Its Cell Cycle Inhibitory, Pro-Apoptotic and Anti-Inflammatory Activities on Human Tumor Cells
A NOVEL RESVERATROL ANALOG : ITS CELL CYCLE INHIBITORY, PRO-APOPTOTIC AND ANTI-INFLAMMATORY ACTIVITIES ON HUMAN TUMOR CELLS A dissertation submitted to Kent State University in partial fulfillment of the requirements for the degree of Doctor of Philosophy by Boren Lin May 2006 Dissertation written by Boren Lin B.S., Tunghai University, 1996 M.S., Kent State University, 2003 Ph. D., Kent State University, 2006 Approved by Dr. Chun-che Tsai , Chair, Doctoral Dissertation Committee Dr. Bryan R. G. Williams , Co-chair, Doctoral Dissertation Committee Dr. Johnnie W. Baker , Members, Doctoral Dissertation Committee Dr. James L. Blank , Dr. Bansidhar Datta , Dr. Gail C. Fraizer , Accepted by Dr. Robert V. Dorman , Director, School of Biomedical Sciences Dr. John R. Stalvey , Dean, College of Arts and Sciences ii TABLE OF CONTENTS LIST OF FIGURES……………………………………………………………….………v LIST OF TABLES……………………………………………………………………….vii ACKNOWLEDGEMENTS….………………………………………………………….viii I INTRODUCTION….………………………………………………….1 Background and Significance……………………………………………………..1 Specific Aims………………………………………………………………………12 II MATERIALS AND METHODS.…………………………………………….16 Cell Culture and Compounds…….……………….…………………………….….16 MTT Cell Viability Assay………………………………………………………….16 Trypan Blue Exclusive Assay……………………………………………………...18 Flow Cytometry for Cell Cycle Analysis……………..……………....……………19 DNA Fragmentation Assay……………………………………………...…………23 Caspase-3 Activity Assay………………………………...……….….…….………24 Annexin V-FITC Staining Assay…………………………………..…...….………28 NF-kappa B p65 Activity Assay……………………………………..………….…29 -

Personalized Prediction of Acquired Resistance to EGFR-Targeted Inhibitors Using a Pathway-Based Machine Learning Approach
Cancers 2019, 11, x S1 of S9 Supplementary Materials: Personalized Prediction of Acquired Resistance to EGFR-Targeted Inhibitors Using a Pathway-Based Machine Learning Approach Young Rae Kim, Yong Wan Kim, Suh Eun Lee, Hye Won Yang and Sung Young Kim Table S1. Characteristics of individual studies. Sample Size Origin of Cancer Drug Dataset Platform S AR (Cell Lines) Lung cancer Agilent-014850 Whole Human GSE34228 26 26 (PC9) Genome Microarray 4x44K Gefitinib Epidermoid carcinoma Affymetrix Human Genome U133 GSE10696 3 3 (A431) Plus 2.0 Head and neck cancer Illumina HumanHT-12 V4.0 GSE62061 12 12 (Cal-27, SSC-25, FaDu, expression beadchip SQ20B) Erlotinib Head and neck cancer Illumina HumanHT-12 V4.0 GSE49135 3 3 (HN5) expression beadchip Lung cancer (HCC827, Illumina HumanHT-12 V3.0 GSE38310 3 6 ER3, T15-2) expression beadchip Lung cancer Illumina HumanHT-12 V3.0 GSE62504 1 2 (HCC827) expression beadchip Afatinib Lung cancer * Illumina HumanHT-12 V4.0 GSE75468 1 3 (HCC827) expression beadchip Head and neck cancer Affymetrix Human Genome U133 Cetuximab GSE21483 3 3 (SCC1) Plus 2.0 Array GEO, gene expression omnibus; GSE, gene expression series; S, sensitive; AR, acquired EGFR-TKI resistant; * Lung Cancer Cells Derived from Tumor Xenograft Model. Table S2. The performances of four penalized regression models. Model ACC precision recall F1 MCC AUROC BRIER Ridge 0.889 0.852 0.958 0.902 0.782 0.964 0.129 Lasso 0.944 0.957 0.938 0.947 0.889 0.991 0.042 Elastic Net 0.978 0.979 0.979 0.979 0.955 0.999 0.023 EPSGO Elastic Net 0.989 1.000 0.979 0.989 0.978 1.000 0.018 AUROC, area under curve of receiver operating characteristic; ACC, accuracy; MCC, Matthews correlation coefficient; EPSGO, Efficient Parameter Selection via Global Optimization algorithm. -
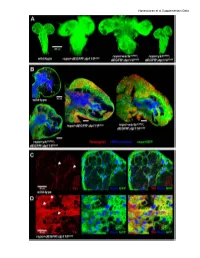
Supplementary Data Vigneswaran Et Al Supplementary Data
Vigneswaran et al Supplementary Data Vigneswaran et al Supplementary Data Figure S1: Yki is required for EGFR-PI3K-driven glial neoplasia in Drosophila (A) Optical projections of whole brain-nerve cord complexes from 3rd instar larvae approximately 130 hrs old. Dorsal view; anterior up. CD8-GFP (green) labels glial cell bodies. Compared to repo>dEGFRλ;dp110CAAX, warts knockdown (repo>wartsdsRNA; dEGFRλ;dp110CAAX) increased neoplastic brain overgrowth and yki knockdown (repo>ykidsRNA;dEGFRλ;dp110CAAX) decreased neoplastic brain overgrowth. (B) 3 µm optical projections of brain hemispheres, age-matched 3rd instar larvae. Frontal sections; anterior up; midline to left. Repo (red) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies; anti-HRP (blue) counter-stains for neurons and neuropil. (middle) repo>dEGFRλ;dp110CAAX showed increased glial cell numbers (red nuclei) compared to (upper left) wild-type. Compared to repo>dEGFRλ;dp110CAAX, (right) warts knockdown increased neoplastic glial cell numbers (red nuclei), whereas (lower left) yki knockdown reduced neoplastic glial cell numbers (red nuclei). (C, D) Low levels of Yki protein (red) was observed in wild-type central brain glia (white arrows, left panel in C) compared to high levels of cytoplasmic and nuclear Yki protein in dEGFRλ;dp110CAAX neoplastic glia (white arrows, left panel in D); Repo (blue) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies. Vigneswaran et al Supplementary Data Figure S2: YAP/TAZ expression confined to RTK-amplified tuMor cells and Maintained in patient-derived xenografts (A) On the left, immunohistochemical (IHC) staining in representative normal brain parenchyma in the cortex where YAP expression and TAZ expression was limited to vascular cells and was not detectable in normal neuronal and glial cells.