DNAJB1–PRKACA Fusion Kinase Interacts with Β-Catenin and the Liver
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Tumour-Agnostic Therapy for Pancreatic Cancer and Biliary Tract Cancer
diagnostics Review Tumour-Agnostic Therapy for Pancreatic Cancer and Biliary Tract Cancer Shunsuke Kato Department of Clinical Oncology, Juntendo University Graduate School of Medicine, 2-1-1, Hongo, Bunkyo-ku, Tokyo 113-8421, Japan; [email protected]; Tel.: +81-3-5802-1543 Abstract: The prognosis of patients with solid tumours has remarkably improved with the develop- ment of molecular-targeted drugs and immune checkpoint inhibitors. However, the improvements in the prognosis of pancreatic cancer and biliary tract cancer is delayed compared to other carcinomas, and the 5-year survival rates of distal-stage disease are approximately 10 and 20%, respectively. How- ever, a comprehensive analysis of tumour cells using The Cancer Genome Atlas (TCGA) project has led to the identification of various driver mutations. Evidently, few mutations exist across organs, and basket trials targeting driver mutations regardless of the primary organ are being actively conducted. Such basket trials not only focus on the gate keeper-type oncogene mutations, such as HER2 and BRAF, but also focus on the caretaker-type tumour suppressor genes, such as BRCA1/2, mismatch repair-related genes, which cause hereditary cancer syndrome. As oncogene panel testing is a vital approach in routine practice, clinicians should devise a strategy for improved understanding of the cancer genome. Here, the gene mutation profiles of pancreatic cancer and biliary tract cancer have been outlined and the current status of tumour-agnostic therapy in these cancers has been reported. Keywords: pancreatic cancer; biliary tract cancer; targeted therapy; solid tumours; driver mutations; agonist therapy Citation: Kato, S. Tumour-Agnostic Therapy for Pancreatic Cancer and 1. -

Identification of TPD52 and DNAJB1 As Two Novel Bile Biomarkers for Cholangiocarcinoma by Itraq‑Based Quantitative Proteomics Analysis
2622 ONCOLOGY REPORTS 42: 2622-2634, 2019 Identification of TPD52 and DNAJB1 as two novel bile biomarkers for cholangiocarcinoma by iTRAQ‑based quantitative proteomics analysis HONGYUE REN1*, MINGXU LUO2,3*, JINZHONG CHEN4, YANMING ZHOU5, XIUMEI LI4, YANYAN ZHAN6, DONGYAN SHEN7 and BO CHEN3 1Department of Pathology, The Affiliated Southeast Hospital of Xiamen University, Zhangzhou, Fujian 363000; 2Department of Gastrointestinal Surgery, Xiamen Humanity Hospital; Departments of 3Gastrointestinal Surgery, 4Endoscopy Center and 5Hepatopancreatobiliary Surgery, Xiamen Cancer Hospital, The First Affiliated Hospital of Xiamen University, Xiamen Fujian 361003; 6Cancer Research Center, Xiamen University Medical College, Xiamen, Fujian 361002; 7Biobank, Xiamen Cancer Hospital, The First Affiliated Hospital of Xiamen University, Xiamen, Fujian 361003, P.R. China Received December 17, 2018; Accepted September 26, 2019 DOI: 10.3892/or.2019.7387 Abstract. Cholangiocarcinoma (CCA) represents a type of proteins may contribute to tumor pathogenesis. In addition, the epithelial cancer with a late diagnosis and poor outcome. expression levels of TPD52 and DNAJB1 were found to be However, the molecular mechanisms responsible for the devel- closely associated with the clinical parameters and prognosis opment of CCA have not yet been fully identified. Thus, in this of patients with CCA. On the whole, the findings of this study study, we aimed to elucidate some of these mechanisms. For indicate that TPD52 and DNAJB1 may serve as novel bile this purpose, isobaric tags for relative and absolute quantifica- biomarkers for CCA. tion (iTRAQ) was performed to analyze the secretory proteins from the 2 CCA cell lines, TFK1 and HuCCT1, as well as from Introduction a normal biliary epithelial cell line, human intrahepatic biliary epithelial cells (HiBECs). -
![Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]](https://docslib.b-cdn.net/cover/8283/computational-genome-wide-identification-of-heat-shock-protein-genes-in-the-bovine-genome-version-1-peer-review-2-approved-1-approved-with-reservations-88283.webp)
Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]
F1000Research 2018, 7:1504 Last updated: 08 AUG 2021 RESEARCH ARTICLE Computational genome-wide identification of heat shock protein genes in the bovine genome [version 1; peer review: 2 approved, 1 approved with reservations] Oyeyemi O. Ajayi1,2, Sunday O. Peters3, Marcos De Donato2,4, Sunday O. Sowande5, Fidalis D.N. Mujibi6, Olanrewaju B. Morenikeji2,7, Bolaji N. Thomas 8, Matthew A. Adeleke 9, Ikhide G. Imumorin2,10,11 1Department of Animal Breeding and Genetics, Federal University of Agriculture, Abeokuta, Nigeria 2International Programs, College of Agriculture and Life Sciences, Cornell University, Ithaca, NY, 14853, USA 3Department of Animal Science, Berry College, Mount Berry, GA, 30149, USA 4Departamento Regional de Bioingenierias, Tecnologico de Monterrey, Escuela de Ingenieria y Ciencias, Queretaro, Mexico 5Department of Animal Production and Health, Federal University of Agriculture, Abeokuta, Nigeria 6Usomi Limited, Nairobi, Kenya 7Department of Animal Production and Health, Federal University of Technology, Akure, Nigeria 8Department of Biomedical Sciences, Rochester Institute of Technology, Rochester, NY, 14623, USA 9School of Life Sciences, University of KwaZulu-Natal, Durban, 4000, South Africa 10School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, 30032, USA 11African Institute of Bioscience Research and Training, Ibadan, Nigeria v1 First published: 20 Sep 2018, 7:1504 Open Peer Review https://doi.org/10.12688/f1000research.16058.1 Latest published: 20 Sep 2018, 7:1504 https://doi.org/10.12688/f1000research.16058.1 Reviewer Status Invited Reviewers Abstract Background: Heat shock proteins (HSPs) are molecular chaperones 1 2 3 known to bind and sequester client proteins under stress. Methods: To identify and better understand some of these proteins, version 1 we carried out a computational genome-wide survey of the bovine 20 Sep 2018 report report report genome. -

Serum Albumin OS=Homo Sapiens
Protein Name Cluster of Glial fibrillary acidic protein OS=Homo sapiens GN=GFAP PE=1 SV=1 (P14136) Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 Cluster of Isoform 3 of Plectin OS=Homo sapiens GN=PLEC (Q15149-3) Cluster of Hemoglobin subunit beta OS=Homo sapiens GN=HBB PE=1 SV=2 (P68871) Vimentin OS=Homo sapiens GN=VIM PE=1 SV=4 Cluster of Tubulin beta-3 chain OS=Homo sapiens GN=TUBB3 PE=1 SV=2 (Q13509) Cluster of Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 (P60709) Cluster of Tubulin alpha-1B chain OS=Homo sapiens GN=TUBA1B PE=1 SV=1 (P68363) Cluster of Isoform 2 of Spectrin alpha chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTAN1 (Q13813-2) Hemoglobin subunit alpha OS=Homo sapiens GN=HBA1 PE=1 SV=2 Cluster of Spectrin beta chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTBN1 PE=1 SV=2 (Q01082) Cluster of Pyruvate kinase isozymes M1/M2 OS=Homo sapiens GN=PKM PE=1 SV=4 (P14618) Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 Clathrin heavy chain 1 OS=Homo sapiens GN=CLTC PE=1 SV=5 Filamin-A OS=Homo sapiens GN=FLNA PE=1 SV=4 Cytoplasmic dynein 1 heavy chain 1 OS=Homo sapiens GN=DYNC1H1 PE=1 SV=5 Cluster of ATPase, Na+/K+ transporting, alpha 2 (+) polypeptide OS=Homo sapiens GN=ATP1A2 PE=3 SV=1 (B1AKY9) Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 Dihydropyrimidinase-related protein 2 OS=Homo sapiens GN=DPYSL2 PE=1 SV=1 Cluster of Alpha-actinin-1 OS=Homo sapiens GN=ACTN1 PE=1 SV=2 (P12814) 60 kDa heat shock protein, mitochondrial OS=Homo -

DNAJB1-PRKACA in HEK293T Cells Induces LINC00473 Overexpression That Depends on PKA Signaling Stephanie S
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.11.455931; this version posted August 11, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. DNAJB1-PRKACA in HEK293T cells induces LINC00473 overexpression that depends on PKA signaling Stephanie S. Kim1*, Ina Kycia1*, Michael Karski1#, Rosanna K. Ma2#, Evan A. Bordt3, Julian Kwan4, Anju Karki1, Elle Winter1, Ranan G. Aktas1, Yuxuan Wu5, Andrew Emili4, Daniel E. Bauer5, Praveen Sethupathy2, Khashayar Vakili1 1. Department of Surgery, Boston Children’s Hospital, Boston, MA, USA 2. Department of Biomedical Sciences, College of Veterinary Medicine, Cornell University, Ithaca, NY, USA 3. Department of Pediatrics, Lurie Center for Autism, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA 4. Center for Networks Systems Biology, Department of Biochemistry, Boston University School of Medicine, 71 E Concord St, Boston MA 02118 5. Division of Hematology/Oncology, Boston Children’s Hospital, Department of Pediatric Oncology, Dana-Farber Cancer Institute, Harvard Stem Cell Institute, Broad Institute, Department of Pediatrics, Harvard Medical School, Boston, MA, USA (*,# -contributed equally to the manuscript) Corresponding Author: Khashayar Vakili, MD Department of Surgery Boston Children’s Hospital 300 Longwood Avenue Boston, MA 02115 Tel: 617-355-8544 [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.08.11.455931; this version posted August 11, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Fibrolamellar carcinoma (FLC) is a primary liver cancer that most commonly arises in adolescents and young adults in a background of normal liver tissue and has an poor prognosis due to lack of effective chemotherapeutic agents. -

PRKACA Mediates Resistance to HER2-Targeted Therapy in Breast Cancer Cells and Restores Anti-Apoptotic Signaling
Oncogene (2015) 34, 2061–2071 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc ORIGINAL ARTICLE PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling SE Moody1,2,3, AC Schinzel1, S Singh1, F Izzo1, MR Strickland1, L Luo1,2, SR Thomas3, JS Boehm3, SY Kim4, ZC Wang5,6 and WC Hahn1,2,3 Targeting HER2 with antibodies or small molecule inhibitors in HER2-positive breast cancer leads to improved survival, but resistance is a common clinical problem. To uncover novel mechanisms of resistance to anti-HER2 therapy in breast cancer, we performed a kinase open reading frame screen to identify genes that rescue HER2-amplified breast cancer cells from HER2 inhibition or suppression. In addition to multiple members of the MAPK (mitogen-activated protein kinase) and PI3K (phosphoinositide 3-kinase) signaling pathways, we discovered that expression of the survival kinases PRKACA and PIM1 rescued cells from anti-HER2 therapy. Furthermore, we observed elevated PRKACA expression in trastuzumab-resistant breast cancer samples, indicating that this pathway is activated in breast cancers that are clinically resistant to trastuzumab-containing therapy. We found that neither PRKACA nor PIM1 restored MAPK or PI3K activation after lapatinib or trastuzumab treatment, but rather inactivated the pro-apoptotic protein BAD, the BCl-2-associated death promoter, thereby permitting survival signaling through BCL- XL. Pharmacological blockade of BCL-XL/BCL-2 partially abrogated the rescue effects conferred by PRKACA and PIM1, and sensitized cells to lapatinib treatment. These observations suggest that combined targeting of HER2 and the BCL-XL/BCL-2 anti-apoptotic pathway may increase responses to anti-HER2 therapy in breast cancer and decrease the emergence of resistant disease. -

Celastrol Increases Glucocerebrosidase Activity in Gaucher Disease by Modulating Molecular Chaperones
Celastrol increases glucocerebrosidase activity in Gaucher disease by modulating molecular chaperones Chunzhang Yanga,1, Cody L. Swallowsa, Chao Zhanga, Jie Lua, Hongbin Xiaob, Roscoe O. Bradya,1, and Zhengping Zhuanga,1 aSurgical Neurology Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892-1260; and bInstitute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China Contributed by Roscoe O. Brady, November 19, 2013 (sent for review October 21, 2013) Gaucher disease is caused by mutations in the glucosidase, beta, acid increased the catalytic activity of mutant GCase. Celastrol interfered gene that encodes glucocerebrosidase (GCase). Glucosidase, beta, with the recruitment of Cdc37 to Hsp90 halting the assembly of the acid mutations often cause protein misfolding and quantitative loss requisite chaperone complex. Inhibition of Hsp90 reduced its rec- of GCase. In the present study, we found that celastrol, an herb de- ognition of mutant GCase and therefore limited the proteasomal rivative with known anticancer, anti-inflammatory, and antioxidant degradation of the mutant protein. Additionally, celastrol triggered activity, significantly increased the quantity and catalytic activity of a reorganization of the gene expression pattern of molecular chap- GCase. Celastrol interfered with the establishment of the heat-shock erones such as DnaJ homolog subfamily B members 1 and 9 protein 90/Hsp90 cochaperone Cdc37/Hsp90-Hsp70-organizing pro- (DNAJB1/9), heat shock 70kDa proteins 1A and 1B (HSPA1A/B), tein chaperone complex with mutant GCase and reduced heat-shock and Bcl2-associated athanogene 3 (BAG3). The presence of BAG protein 90-associated protein degradation. In addition, celastrol mod- family molecular chaperone regulator 3 (BAG3) further stabi- ulated the expression of molecular chaperones. -

Personalized Prediction of Acquired Resistance to EGFR-Targeted Inhibitors Using a Pathway-Based Machine Learning Approach
Cancers 2019, 11, x S1 of S9 Supplementary Materials: Personalized Prediction of Acquired Resistance to EGFR-Targeted Inhibitors Using a Pathway-Based Machine Learning Approach Young Rae Kim, Yong Wan Kim, Suh Eun Lee, Hye Won Yang and Sung Young Kim Table S1. Characteristics of individual studies. Sample Size Origin of Cancer Drug Dataset Platform S AR (Cell Lines) Lung cancer Agilent-014850 Whole Human GSE34228 26 26 (PC9) Genome Microarray 4x44K Gefitinib Epidermoid carcinoma Affymetrix Human Genome U133 GSE10696 3 3 (A431) Plus 2.0 Head and neck cancer Illumina HumanHT-12 V4.0 GSE62061 12 12 (Cal-27, SSC-25, FaDu, expression beadchip SQ20B) Erlotinib Head and neck cancer Illumina HumanHT-12 V4.0 GSE49135 3 3 (HN5) expression beadchip Lung cancer (HCC827, Illumina HumanHT-12 V3.0 GSE38310 3 6 ER3, T15-2) expression beadchip Lung cancer Illumina HumanHT-12 V3.0 GSE62504 1 2 (HCC827) expression beadchip Afatinib Lung cancer * Illumina HumanHT-12 V4.0 GSE75468 1 3 (HCC827) expression beadchip Head and neck cancer Affymetrix Human Genome U133 Cetuximab GSE21483 3 3 (SCC1) Plus 2.0 Array GEO, gene expression omnibus; GSE, gene expression series; S, sensitive; AR, acquired EGFR-TKI resistant; * Lung Cancer Cells Derived from Tumor Xenograft Model. Table S2. The performances of four penalized regression models. Model ACC precision recall F1 MCC AUROC BRIER Ridge 0.889 0.852 0.958 0.902 0.782 0.964 0.129 Lasso 0.944 0.957 0.938 0.947 0.889 0.991 0.042 Elastic Net 0.978 0.979 0.979 0.979 0.955 0.999 0.023 EPSGO Elastic Net 0.989 1.000 0.979 0.989 0.978 1.000 0.018 AUROC, area under curve of receiver operating characteristic; ACC, accuracy; MCC, Matthews correlation coefficient; EPSGO, Efficient Parameter Selection via Global Optimization algorithm. -
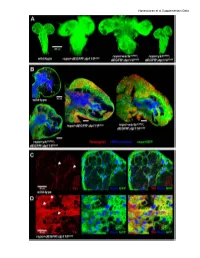
Supplementary Data Vigneswaran Et Al Supplementary Data
Vigneswaran et al Supplementary Data Vigneswaran et al Supplementary Data Figure S1: Yki is required for EGFR-PI3K-driven glial neoplasia in Drosophila (A) Optical projections of whole brain-nerve cord complexes from 3rd instar larvae approximately 130 hrs old. Dorsal view; anterior up. CD8-GFP (green) labels glial cell bodies. Compared to repo>dEGFRλ;dp110CAAX, warts knockdown (repo>wartsdsRNA; dEGFRλ;dp110CAAX) increased neoplastic brain overgrowth and yki knockdown (repo>ykidsRNA;dEGFRλ;dp110CAAX) decreased neoplastic brain overgrowth. (B) 3 µm optical projections of brain hemispheres, age-matched 3rd instar larvae. Frontal sections; anterior up; midline to left. Repo (red) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies; anti-HRP (blue) counter-stains for neurons and neuropil. (middle) repo>dEGFRλ;dp110CAAX showed increased glial cell numbers (red nuclei) compared to (upper left) wild-type. Compared to repo>dEGFRλ;dp110CAAX, (right) warts knockdown increased neoplastic glial cell numbers (red nuclei), whereas (lower left) yki knockdown reduced neoplastic glial cell numbers (red nuclei). (C, D) Low levels of Yki protein (red) was observed in wild-type central brain glia (white arrows, left panel in C) compared to high levels of cytoplasmic and nuclear Yki protein in dEGFRλ;dp110CAAX neoplastic glia (white arrows, left panel in D); Repo (blue) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies. Vigneswaran et al Supplementary Data Figure S2: YAP/TAZ expression confined to RTK-amplified tuMor cells and Maintained in patient-derived xenografts (A) On the left, immunohistochemical (IHC) staining in representative normal brain parenchyma in the cortex where YAP expression and TAZ expression was limited to vascular cells and was not detectable in normal neuronal and glial cells. -

Dependent Protein Kinase (PRKACA) Gene in Japanese Patients with Several Adrenal Adenomas Secreting Cortisol
Endocrine Journal 2014, 61 (8), 825-832 RAPID COMMUNICATION Somatic mutations of the catalytic subunit of cyclic AMP- dependent protein kinase (PRKACA) gene in Japanese patients with several adrenal adenomas secreting cortisol Yasuyo Nakajima1)*, Takashi Okamura1)*, Tamae Gohko1), Tetsurou Satoh1), Koshi Hashimoto1), Nobuyuki Shibusawa1), Atsushi Ozawa1), Sumiyasu Ishii1), Takuya Tomaru1), Kazuhiko Horiguchi1), Shuichi Okada1), Daisuke Takata2), Nana Rokutanda2), Jun Horiguchi2), Yoshito Tsushima3), Tetsunari Oyama4), Izumi Takeyoshi2) and Masanobu Yamada1) 1) Department of Medicine and Molecular Science, Gunma University Graduate School of Medicine, Maebashi 371-8511, Japan 2) Department of Thoracic and Visceral Organ Surgery, Gunma University Graduate School of Medicine, Maebashi 371-8511, Japan 3) Department of Diagnostic Radiology and Nuclear Medicine, Gunma University Graduate School of Medicine, Maebashi 371- 8511, Japan 4) Department of Diagnostic Pathology, Gunma University Graduate School of Medicine, Maebashi 371-8511, Japan Abstract. Somatic mutations of the catalytic subunit of the cyclic AMP-dependent protein kinase (PRKACA) gene have recently been identified in about 35% of cortisol-producing adenomas (CPAs), with the affected patients showing overt Cushing’s syndrome. Since we recently reported higher prevalence of mutations of the KCNJ5 gene and associations with autonomous cortisol secretion in Japanese aldosterone-producing adenomas than in Western countries, there might be different features of CPAs between Japan and the West. We therefore investigated mutations of the PRKACA gene in Japanese patients with several adrenal tumors secreting cortisol, including overt Cushing’s syndrome, subclinical Cushing’s syndrome, and aldosterone-producing adenomas (APAs) co-secreting cortisol operated on at Gunma University Hospital. Of the 13 patients with CPA who showed overt Cushing’s syndrome, 3 (23%) had recurrent somatic mutations of the PRKACA gene, p.L206R (c.617 T>G), and there were no mutations in subclinical Cushing’s syndrome.