DBV TECHNOLOGIES S.A. (Exact Name of Registrant As Specified in Its Charter and Translation of Registrant’S Name Into English)
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
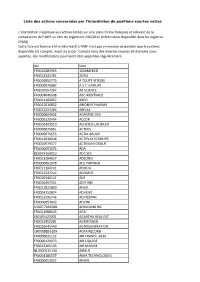
Liste Des Actions Concernées Par L'interdiction De Positions Courtes Nettes
Liste des actions concernées par l'interdiction de positions courtes nettes L’interdiction s’applique aux actions listées sur une plate-forme française et relevant de la compétence de l’AMF au titre du règlement 236/2012 (information disponible dans les registres ESMA). Cette liste est fournie à titre informatif. L'AMF n'est pas en mesure de garantir que le contenu disponible est complet, exact ou à jour. Compte tenu des diverses sources de données sous- jacentes, des modifications pourraient être apportées régulièrement. Isin Nom FR0010285965 1000MERCIS FR0013341781 2CRSI FR0010050773 A TOUTE VITESSE FR0000076887 A.S.T. GROUPE FR0010557264 AB SCIENCE FR0004040608 ABC ARBITRAGE FR0013185857 ABEO FR0012616852 ABIONYX PHARMA FR0012333284 ABIVAX FR0000064602 ACANTHE DEV. FR0000120404 ACCOR FR0010493510 ACHETER-LOUER.FR FR0000076861 ACTEOS FR0000076655 ACTIA GROUP FR0011038348 ACTIPLAY (GROUPE) FR0010979377 ACTIVIUM GROUP FR0000053076 ADA BE0974269012 ADC SIIC FR0013284627 ADEUNIS FR0000062978 ADL PARTNER FR0011184241 ADOCIA FR0013247244 ADOMOS FR0010340141 ADP FR0010457531 ADTHINK FR0012821890 ADUX FR0004152874 ADVENIS FR0013296746 ADVICENNE FR0000053043 ADVINI US00774B2088 AERKOMM INC FR0011908045 AG3I ES0105422002 AGARTHA REAL EST FR0013452281 AGRIPOWER FR0010641449 AGROGENERATION CH0008853209 AGTA RECORD FR0000031122 AIR FRANCE -KLM FR0000120073 AIR LIQUIDE FR0013285103 AIR MARINE NL0000235190 AIRBUS FR0004180537 AKKA TECHNOLOGIES FR0000053027 AKWEL FR0000060402 ALBIOMA FR0013258662 ALD FR0000054652 ALES GROUPE FR0000053324 ALPES (COMPAGNIE) -

Document De Référence 2014 Document De Référence
DOCUMENT DE RÉFÉRENCE 2014 DBV TECHNOLOGIES 2014 DOCUMENT DE RÉFÉRENCE DOCUMENT 2014 DE RÉFÉRENCE DBV TECHNOLOGIES Société Anonyme au capital de 1 966 166,10 euros Green Square Bâtiment D, 80/84 rue des Meuniers 92220 Bagneux 441 772 522 R.C.S. Nanterre En application de son règlement général, notamment de l’article 212-13, l’Autorité des marchés financiers a enregistré le présent document de référence le 02 juillet 2015 sous le numéro R.15-057. Ce document ne peut être utilisé à l’appui d’une opération financière que s’il est complété par une note d’opération visée par l’AMF. Il a été établi par l’émetteur et engage la responsabilité de ses signataires. L’enregistrement, conformément aux dispositions de l’article L. 621-8-1-I du code monétaire et financier, a été effectué après que l’AMF a vérifié que le document est complet et compréhensible et que les informations qu’il contient sont cohérentes. Il n’implique pas l’authentification par l’AMF des éléments comptables et financiers présentés. Incorporation par référence : En application de l’article 28 du règlement européen 809/2004, les éléments suivants sont inclus par référence dans le présent document de référence : • Les comptes annuels établis conformément aux principes comptables français au 31 décembre 2013, les comptes établis selon le référentiel IFRS tel qu’adoptés dans l’Union européenne au 31 décembre 2013, et les rapports des commissaires aux comptes y afférents, présentés respectivement aux pages 153 à 190, 191 à 208, 209 et 210-211 du documents de référence n° R.14-017 enregistré par l’Autorités des marchés financiers le 16 avril 2014 ; • Les comptes annuels établis conformément aux principes comptables français au 31 décembre 2012, les comptes établis selon le référentiel IFRS tel qu’adoptés dans l’Union européenne au 31 décembre 2012, et les rapports des commissaires aux comptes y afférents, présentés respectivement aux pages 134 à 162, 163 à 178, 180, 181-182 du document de référence n° R.13-015 enregistré par l’Autorité des marchés financiers le 24 avril 2013. -

Putnam Panagora Market Neutral Fund Q3 Portfolio Holdings
Putnam PanAgora Market Neutral Fund The fund's portfolio 5/31/20 (Unaudited) INVESTMENT COMPANIES (46.1%)(a) Shares Value Morgan Stanley Emerging Markets Domestic Debt Fund, Inc. 640 $3,635 State Street Institutional U.S. Government Money Market Fund 3,939,067 3,939,067 Total investment companies (cost $3,943,561) $3,942,702 UNITS (11.0%)(a) Units Value Acamar Partners Acquisition Corp.(NON) 419 $4,291 Alussa Energy Acquisition Corp. (Cayman Islands)(NON) 856 8,483 Amplitude Healthcare Acquisition Corp.(NON) 2,947 29,529 B. Riley Principal Merger Corp. II(NON) 2,620 26,174 CC Neuberger Principal Holdings I(NON) 2,652 27,024 Chardan Healthcare Acquisition 2 Corp.(NON) 2,652 26,493 CHP Merger Corp.(NON) 2,747 27,745 CIIG Merger Corp.(NON) 4,529 45,335 Collective Growth Corp.(NON) 2,803 27,890 DFP Healthcare Acquisitions Corp.(NON) 2,866 28,746 dMY Technology Group, Inc.(NON) 2,885 29,196 East Stone Acquisition Corp.(NON) 4,230 42,089 FinServ Acquisition Corp.(NON) 831 8,194 Foley Trasimene Acquisition Corp.(NON) 2,626 26,917 Fortress Value Acquisition Corp.(NON) 2,652 26,547 Galileo Acquisition Corp.(NON) 888 8,827 GigCapital3, Inc.(NON) 2,833 28,160 Gores Holdings IV, Inc.(NON) 1,306 13,844 Greenrose Acquisition Corp.(NON) 3,350 32,931 GX Acquisition Corp.(NON) 417 4,233 Healthcare Merger Corp.(NON) 2,705 28,105 InterPrivate Acquisition Corp.(NON) 2,918 29,180 Jaws Acquisition Corp.(NON) 2,620 27,038 Juniper Industrial Holdings, Inc.(NON) 841 8,418 Landcadia Holdings II, Inc.(NON) 1,165 12,174 LGL Systems Acquisition Corp.(NON) 2,568 25,629 Lifesci Acquisition Corp.(NON) 2,866 29,806 LIV Capital Acquisition Corp. -

ESMA Report on Application of AMP 2020
Report To the European Commission on the application of accepted market practices 16 December 2020 | ESMA70-156-3782 ESMA REGULAR USE Table of Contents 1 Executive Summary ....................................................................................................... 2 2 Background .................................................................................................................... 3 3 Information on the legal situation of AMPs established under MAD and AMPs established under MAR ............................................................................................................................ 5 3.1 CNMV ..................................................................................................................... 5 3.2 CMVM ..................................................................................................................... 6 3.3 CONSOB ................................................................................................................ 6 3.4 AMF ........................................................................................................................ 6 4 Application of the established AMPs............................................................................... 7 4.1 AMP established by the CNMV under MAR ............................................................ 7 4.2 AMP established by the CMVM under MAR ............................................................ 8 4.3 AMP established by CONSOB under MAD and MAR ............................................. -

Structures De Gouvernance Des Sociétés Cotées Radiographie
Structures de gouvernance des sociétés cotées Radiographie Observatoire Capital humain Centre de Gouvernement d'Entreprise Octobre 2015 Second line optional lorem ipsum B Subhead lorem ipsum, date quatueriure Mesurer pour comprendre, comprendre pour agir, agir pour progresser Sommaire Executive Summary 2 Préambule 4 1. Radiographie des conseils 7 Introduction 7 Typologie des conseils 8 Indépendance des administrateurs 12 Féminisation des conseils 14 Internationalisation des conseils 18 Distribution par âge et par ancienneté 20 Cumul des mandats 22 Mercato 2014-2015 25 Rémunérations des administrateurs 29 Evaluation des conseils 38 2. Les comités du Conseil 42 Introduction 43 Comités spécialisés 45 3. Assemblées générales 2015 52 Introduction 53 Nature des résolutions 54 Résultats des votes 56 Facteurs d'influence 66 Liste des sociétés 70 Structures de gouvernance des sociétés cotées - Radiographie 1 Executive Summary Des pratiques globalement en ligne avec les codes de gouvernance, mais des différences marquées selon l'indice de cotation Nous sommes très heureux de vous présenter la Nous étudierons les comités spécialisés, leur fréquence radiographie de la gouvernance des sociétés cotées et leur composition, ainsi que la distribution des rôles 2015, issue de l’analyse des pratiques de gouvernement des administrateurs en leur sein. d’entreprise de plus de 300 sociétés cotées au marché Le chapitre relatif aux assemblées générales 2015 Euronext de la Bourse de Paris. analysera près de 5 700 résolutions présentées lors Le gouvernement d’entreprise désigne les modes de 318 assemblées générales en rapprochant les taux de fonctionnement des organes d'administration et d’approbation, la nature des résolutions, la structure de surveillance d’une organisation et leurs relations actionnariale des sociétés et les recommandations de avec la direction et les actionnaires. -

France FTT List of Eligible Companies
ISIN Code Security Name Effective Date FR0000120404 ACCOR August 1, 2012 US00435F3091 ACCOR SPONSORED ADR March 2, 2015 FR0010340141 AEROPORTS DE PARIS August 1, 2012 US0091191082 AIR FRANCE - KLM ADR SPONSORED December 1, 2012 FR0000031122 AIR FRANCE-KLM August 1, 2012 FR0000120073 AIR LIQUIDE August 1, 2012 US0091262024 AIR LIQUIDE ADR December 1, 2012 FR0000130007 ALCATEL LUCENT August 1, 2012 US0212442075 ALSTOM ADR December 1, 2012 FR0010220475 ALSTOM REGROUPT August 1, 2012 FR0000033219 ALTAREA August 1, 2012 FR0000071946 ALTEN January 1, 2014 US02209U1088 ALTRAN TECHN.ADR SPONSORED January 1, 2014 FR0000034639 ALTRAN TECHNOLOGIES January 1, 2014 FR0004125920 AMUNDI January 1, 2016 FR0011027143 AREVA August 1, 2012 US04012G1022 AREVA ADR UNSPONSORED December 1, 2012 FR0010313833 ARKEMA August 1, 2012 US0412321095 ARKEMA ADR December 1, 2012 FR0000076952 ARTOIS (INDLE ET FIN.) NOM. January 1, 2014 FR0000051732 ATOS August 1, 2012 US04962A1051 ATOS ADR UNSPONSORED December 1, 2012 FR0000120628 AXA August 1, 2012 US0545361075 AXA UAP ADR SPONSORED December 1, 2012 FR0000035164 BENETEAU January 1, 2016 FR0000120966 BIC August 1, 2012 US0887361030 BIC ADR December 1, 2012 FR0010096479 BIOMERIEUX August 1, 2012 US09074E1010 BIOMERIEUX ADR September 17, 2015 FR0000131104 BNP PARIBAS ACTIONS A August 1, 2012 US05565A1034 BNP PARIBAS ADR SPONSORED December 1, 2012 US05565A2024 BNP PARIBAS ADR SPONSORED December 1, 2012 FR0000061129 BOIRON January 1, 2015 FR0000039299 BOLLORE August 1, 2012 FR0000120503 BOUYGUES August 1, 2012 US1021171087 -

Neia 20120906
Index Announcement Issue Date: Thursday 6 September 2012 Effective Date: Monday 24 September 2012 Announcement No: 2012-170 ______________________________________________________________________ Indices: Index ISIN Code CAC 40® FR0003500008 CAC Next 20® QS0010989109 CAC® Large 60 QS0011213657 SBF 120® FR0003999481 CAC® Mid 60 QS0010989117 CAC® Small QS0010989125 CAC® Mid & Small QS0010989133 CAC® All-Tradable FR0003999499 CAC® All-Share Index QS0010989141 Subject: Quarterly Review Index Adjustment: Changes in o compositions, (see appendix); o numbers of shares; o free float factors, (preliminary factors see appendix); o capping factors; o divisors. Conditions: The compiler retains the right to change the published selection in case of mergers, take-overs, suspension or resumption of trading during the period before the effective date of the review. Time table: After close of markets on Wednesday 19 September 2012 the following data will be published for each index and each constituent: o number of shares; o final free float percentage; o capping factor. After close of business on Friday 21 September 2012, the new divisor of the CAC 40 index will be published. Page 1 of 13 Avis Indice Date de publication : Jeudi 6 Septembre 2012 Date d’effet : Lundi 24 Septembre 2012 Numéro d’avis : 2012-170 ______________________________________________________________________ Indices: Indice Code ISIN CAC 40® FR0003500008 CAC Next 20® QS0010989109 CAC® Large 60 QS0011213657 SBF 120® FR0003999481 CAC® Mid 60 QS0010989117 CAC® Small QS0010989125 CAC® Mid & Small QS0010989133 CAC® All-Tradable FR0003999499 CAC® All-Share Index QS0010989141 Sujet: Révision trimestrielle Détails : Modifications o de la composition des indices, (voir l’annexe); o du nombre d’actions; o des flottants, (flottants préliminaires voir l’annexe); o des facteurs de plafonnement; o des diviseurs. -

Expiry Months Listed Per Class
31-May-2021 PARIS INDIVIDUAL EQUITY OPTIONS - AMERICAN STYLE - EXPIRY CYCLES Cycle Expiry Months Cycle Lifetime (Months) Monthly Every Month 1; 2; 3 Quarterly March, June, September, December 6; 9; 12 Half-Yearly June, December 18; 24; 30; 36 GROUP GROUP 1 Yearly December 48; 60 Cycle Expiry Months Cycle Lifetime (Months) Monthly Every Month 1; 2; 3 Quarterly March, June, September, December 6; 9; 12 GROUP GROUP 2 Half-Yearly June, December 18; 24 Cycle Expiry Months Cycle Lifetime (Months) Monthly Every Month 1; 2; 3 GROUP GROUP 3 Quarterly March, June, September, December 6; 9; 12 Cycle Expiry Months Cycle Lifetime (Months) Quarterly March, June, September, December 3; 6; 9; 12 GROUP GROUP 4 Cycle Expiry Months Cycle Lifetime (Months) Monthly Every Month 1; 2; 3 SPOTLIGHT OPTIONS Four separate option classes per underlying value are listed. Each class will have a fixed last day of trading, this is either the first, the second, the fourth or, when applicable, the fifth Friday of the month. Weekly options are available for trading as from the Friday two weeks prior to expiry. WEEKLY OPTIONS PARIS INDIVIDUAL EQUITY OPTIONS - EUROPEAN STYLE - EXPIRY CYCLES Cycle Expiry Months Cycle Lifetime (Months) Monthly Every Month 1; 2; 3 Quarterly March, June, September, December 6; 9; 12 GROUP GROUP 1 Half-Yearly June, December 18; 24 Cycle Expiry Months Cycle Lifetime (Months) Half-Yearly June, December 6; 12; 18; 24 GROUP GROUP 2 Cycle Expiry Months Cycle Lifetime (Months) Half-Yearly June, December 6; 12 GROUP GROUP 3 American Style Option Classes -

DBV Technologies Announces Appointment of Michel De Rosen As Non-Executive Chairman of the Board of Directors
Montrouge, France, March 5, 2019 DBV Technologies Announces Appointment of Michel de Rosen as Non-Executive Chairman of the Board of Directors Mr. de Rosen to succeed DBV’s co-founder, Dr. Pierre-Henri Benhamou, who will retire from the Board of Directors and join Scientific Advisory Board Chief Executive Officer, Daniel Tassé, has also been appointed to the Board of Directors DBV Technologies (Euronext: DBV – ISIN: FR0010417345 - Nasdaq Stock Market: DBVT), a clinical-stage biopharmaceutical company, today announced that the Board of Directors has appointed existing board member, Michel de Rosen, as Non- Executive Chairman of the Board. Mr. de Rosen succeeds DBV Technologies’ co- founder, Dr. Pierre-Henri Benhamou, who retired from his position as DBV’s Non- Executive Chairman and board member. Dr. Benhamou will join the Company’s Scientific Advisory Board, effective immediately. Additionally, Daniel Tassé, Chief Executive Officer of DBV Technologies, has been appointed to the Board, replacing Dr. Pierre-Henri Benhamou. With these changes, effective immediately, DBV’s Board consists of eight directors. “On behalf of DBV’s Board, I would like to thank Pierre-Henri for his leadership and ongoing commitment to this organization. We also welcome Daniel, as well as Michel and his expanded role, and look forward to our continued partnership in building a leading biopharmaceutical company,” said Dr. Torbjörn Bjerke, DBV Technologies Board Director. Mr. de Rosen has served on DBV’s Board of Directors since June 2018. He is currently Chairman of the Board of Faurecia, a global supplier of automotive equipment, and Chairman of the Board of Pharnext, a publicly-traded pharmaceutical company. -

Document De Reference
2015 DOCUMENT DE REFERENCE The epicutaneous immunotherapy company DOCUMENT 2015 DE RÉFÉRENCE DBV TECHNOLOGIES Société Anonyme au capital de 2 420 512,90 euros 177-181 Avenue Pierre-Brossolette 92120 Montrouge 441 772 522 R.C.S. Nanterre Le présent document de référence a été déposé auprès de l’Autorité des marchés financiers le 28 avril 2016 sous le numéro D.16-0437, conformément à l’article 212-13 de son règlement général. Il pourra être utilisé à l’appui d’une opération financière s’il est complété par une note d’opération visée par l’AMF. Ce document a été établi par l’émetteur et engage la responsabilité de ses signataires. Incorporation par référence : En application de l’article 28 du règlement européen 809/2004, les éléments suivants sont inclus par référence dans le présent document de référence : • Les comptes annuels établis conformément aux principes comptables français au 31 décembre 2014, les comptes consolidés établis selon le référentiel IFRS tel qu’adopté par l’Union européenne au 31 décembre 2014, et les rapports des commissaires aux comptes y afférents respectivement aux pages 182 à 195, 143 à 181, 199-200 et 198-199 du document de référence n° R.15-057 déposé auprès de l’Autorité des marchés financiers le 2 juillet 2015. • Les comptes annuels établis conformément aux principes comptables français au 31 décembre 2013, les comptes établis selon le référentiel IFRS tel qu’adoptés par l’Union européenne au 31 décembre 2013, et les rapports des commissaires aux comptes y afférents respectivement aux pages 191 à 208, 153 à 190, 210-211 et 209 du document de référence n° R.14-017 déposé auprès de l’Autorité des marchés financiers le 16 avril 2014. -

Company Overview Valuation Data Source
Valuation Data Source company overview No. Company No. Company No. Company "Bank "Saint-Petersburg" Public 60 AbClon Inc. 117 Activision Blizzard, Inc. 1 Joint-Stock Company Abdullah Al-Othaim Markets 118 Actron Technology Corporation 61 2 1&1 Drillisch AG Company 119 Actuant Corporation 3 1-800-FLOWERS.COM, Inc. Abdulmohsen Al-Hokair Group for 120 Acuity Brands, Inc. 62 4 11 bit studios S.A. Tourism and Development Company 121 Acushnet Holdings Corp. 5 1st Constitution Bancorp 63 Abengoa, S.A. 122 Ad-Sol Nissin Corporation 6 1st Source Corporation 64 Abeona Therapeutics Inc. 123 Adairs Limited 7 21Vianet Group, Inc. 65 Abercrombie & Fitch Co. 124 ADAMA Ltd. 8 22nd Century Group, Inc. 66 Ability Enterprise Co., Ltd. 125 Adamas Pharmaceuticals, Inc. Ability Opto-Electronics Technology 126 Adamis Pharmaceuticals Corporation 9 2U, Inc. 67 Co.,Ltd. 127 Adani Enterprises Limited 10 3-D Matrix, Ltd. 68 Abiomed, Inc. 128 Adani Gas Limited 11 361 Degrees International Limited 69 ABIST Co.,Ltd. 129 Adani Green Energy Limited 12 3D Systems Corporation 70 ABL Bio Inc. Adani Ports and Special Economic 13 3i Group plc 130 71 Able C&C Co., Ltd. Zone Limited 14 3M Company 131 Adani Power Limited 72 ABM Industries Incorporated 15 3M India Limited 132 Adani Transmissions Limited 73 ABN AMRO Bank N.V. 16 3S KOREA Co., Ltd. 133 Adaptimmune Therapeutics plc 74 Aboitiz Equity Ventures, Inc. 17 3SBio Inc. 134 Adastria Co., Ltd. 75 Aboitiz Power Corporation 18 500.com Limited 135 ADATA Technology Co., Ltd. 76 Abraxas Petroleum Corporation 19 51 Credit Card Inc. -

Fidelity® Total International Index Fund
Quarterly Holdings Report for Fidelity® Total International Index Fund July 31, 2021 TI1-QTLY-0921 1.9871048.105 Schedule of Investments July 31, 2021 (Unaudited) Showing Percentage of Net Assets Common Stocks – 96.2% Shares Value Argentina – 0.0% Banco Macro SA sponsored ADR (a) (b) 12,515 $ 168,953 Grupo Financiero Galicia SA sponsored ADR 40,080 313,025 Pampa Holding SA sponsored ADR (a) (b) 14,899 231,083 Transportadora de Gas del Sur SA Class B sponsored ADR (a) (b) 20,041 88,180 YPF SA Class D sponsored ADR (a) 47,032 205,060 TOTAL ARGENTINA 1,006,301 Australia – 4.4% Abacus Property Group unit 107,655 250,438 Accent Group Ltd. 71,378 142,999 Adbri Ltd. 107,008 277,988 Afterpay Ltd. (a) 53,792 3,815,679 AGL Energy Ltd. 147,705 783,684 ALS Ltd. 115,948 1,084,027 Altium Ltd. 30,470 758,018 Alumina Ltd. 658,550 807,072 AMP Ltd. 828,260 632,131 Ampol Ltd. 59,920 1,242,217 Ansell Ltd. 32,944 948,906 APA Group unit 278,390 1,946,946 Appen Ltd. 26,037 217,058 ARB Corp. Ltd. 20,573 705,053 Aristocrat Leisure Ltd. 141,396 4,320,710 ASX Ltd. 50,230 2,834,264 Atlas Arteria Ltd. unit (b) 219,856 1,011,610 Aub Group Ltd. 26,473 447,797 Aurizon Holdings Ltd. 456,059 1,291,861 AusNet Services 479,355 641,989 Austal Ltd. 109,179 173,061 Australia & New Zealand Banking Group Ltd. 705,248 14,341,206 Australian Agricultural Co.