Metabarcoding of Freshwater Invertebrates to Detect the Effects of a Pesticide Spill
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Genomanalyse Von Prodiamesa Olivacea
Genomanalyse von Prodiamesa olivacea Dissertation zur Erlangung des Grades Doktor der Naturwissenschaften (Dr. rer. nat.) am Fachbereich Biologie der Johannes Gutenberg-Universität in Mainz Sarah Brunck geb. 08.08.1987 in Mainz Mainz, 2016 Dekan: 1. Berichterstatter: 2. Berichterstatter: Tag der mündlichen Prüfung: ii Inhaltsverzeichnis Inhaltsverzeichnis ................................................................................................................................ iii 1 Einleitung ........................................................................................................................................... 1 1.1 Die Familie der Chironomiden ................................................................................................. 1 1.1.1 Die Gattung Chironomus ..................................................................................................... 3 1.1.2 Die Gattung Prodiamesa ....................................................................................................... 6 1.2 Die Struktur von Insekten-Genomen am Beispiel der Chironomiden ............................... 9 1.2.1 Hochrepetitive DNA-Sequenzen ..................................................................................... 11 1.2.2 Mittelrepetitive DNA-Sequenzen bzw. Gen-Familien ................................................. 13 1.2.3 Gene und genregulatorische Sequenzen ........................................................................ 17 1.3 Zielsetzung ............................................................................................................................... -

CHIRONOMUS Newsletter on Chironomidae Research
CHIRONOMUS Newsletter on Chironomidae Research No. 25 ISSN 0172-1941 (printed) 1891-5426 (online) November 2012 CONTENTS Editorial: Inventories - What are they good for? 3 Dr. William P. Coffman: Celebrating 50 years of research on Chironomidae 4 Dear Sepp! 9 Dr. Marta Margreiter-Kownacka 14 Current Research Sharma, S. et al. Chironomidae (Diptera) in the Himalayan Lakes - A study of sub- fossil assemblages in the sediments of two high altitude lakes from Nepal 15 Krosch, M. et al. Non-destructive DNA extraction from Chironomidae, including fragile pupal exuviae, extends analysable collections and enhances vouchering 22 Martin, J. Kiefferulus barbitarsis (Kieffer, 1911) and Kiefferulus tainanus (Kieffer, 1912) are distinct species 28 Short Communications An easy to make and simple designed rearing apparatus for Chironomidae 33 Some proposed emendations to larval morphology terminology 35 Chironomids in Quaternary permafrost deposits in the Siberian Arctic 39 New books, resources and announcements 43 Finnish Chironomidae 47 Chironomini indet. (Paratendipes?) from La Selva Biological Station, Costa Rica. Photo by Carlos de la Rosa. CHIRONOMUS Newsletter on Chironomidae Research Editors Torbjørn EKREM, Museum of Natural History and Archaeology, Norwegian University of Science and Technology, NO-7491 Trondheim, Norway Peter H. LANGTON, 16, Irish Society Court, Coleraine, Co. Londonderry, Northern Ireland BT52 1GX The CHIRONOMUS Newsletter on Chironomidae Research is devoted to all aspects of chironomid research and aims to be an updated news bulletin for the Chironomidae research community. The newsletter is published yearly in October/November, is open access, and can be downloaded free from this website: http:// www.ntnu.no/ojs/index.php/chironomus. Publisher is the Museum of Natural History and Archaeology at the Norwegian University of Science and Technology in Trondheim, Norway. -

The 17Th International Colloquium on Amphipoda
Biodiversity Journal, 2017, 8 (2): 391–394 MONOGRAPH The 17th International Colloquium on Amphipoda Sabrina Lo Brutto1,2,*, Eugenia Schimmenti1 & Davide Iaciofano1 1Dept. STEBICEF, Section of Animal Biology, via Archirafi 18, Palermo, University of Palermo, Italy 2Museum of Zoology “Doderlein”, SIMUA, via Archirafi 16, University of Palermo, Italy *Corresponding author, email: [email protected] th th ABSTRACT The 17 International Colloquium on Amphipoda (17 ICA) has been organized by the University of Palermo (Sicily, Italy), and took place in Trapani, 4-7 September 2017. All the contributions have been published in the present monograph and include a wide range of topics. KEY WORDS International Colloquium on Amphipoda; ICA; Amphipoda. Received 30.04.2017; accepted 31.05.2017; printed 30.06.2017 Proceedings of the 17th International Colloquium on Amphipoda (17th ICA), September 4th-7th 2017, Trapani (Italy) The first International Colloquium on Amphi- Poland, Turkey, Norway, Brazil and Canada within poda was held in Verona in 1969, as a simple meet- the Scientific Committee: ing of specialists interested in the Systematics of Sabrina Lo Brutto (Coordinator) - University of Gammarus and Niphargus. Palermo, Italy Now, after 48 years, the Colloquium reached the Elvira De Matthaeis - University La Sapienza, 17th edition, held at the “Polo Territoriale della Italy Provincia di Trapani”, a site of the University of Felicita Scapini - University of Firenze, Italy Palermo, in Italy; and for the second time in Sicily Alberto Ugolini - University of Firenze, Italy (Lo Brutto et al., 2013). Maria Beatrice Scipione - Stazione Zoologica The Organizing and Scientific Committees were Anton Dohrn, Italy composed by people from different countries. -

Field Host Range, Foraging Depth, and Impact Of
FIELD HOST RANGE, FORAGING DEPTH, AND IMPACT OF CRICOTOPUS LEBETIS SUBLETTE (DIPTERA: CHIRONOMIDAE), A BIOLOGICAL CONTROL AGENT OF HYDRILLA VERTICILLATA (L.F.) ROYLE (HYDROCHARITACEAE) By EUTYCHUS MUKURE KARIUKI A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA 2017 © 2017 Eutychus Mukure Kariuki To my loving family ACKNOWLEDGMENTS I would like to thank my Major Advisor, Dr. Raymond L. Hix, and my Co-Advisor, Dr. James P. Cuda, for their support and guidance during my Ph.D. program. I am also thankful to my committee members, Dr. Stephen D. Hight for his invaluable support and mentorship during the course of my research; Dr. Jennifer Gillett-Kaufman for her constant support, especially through the writing process of my dissertation; and Dr. Lyn Gettys for always being available to help with questions. I am grateful to all others who provided their assistance, including Dr. Edzard van Santen (University of Florida), Dr. Lazarus Mramba (University of Florida), Dr. Emma Weeks (University of Florida), John Mass (United States Department of Agriculture (USDA), Tallahassee, Florida), Kelle Sullivan (Florida Fish and Wildlife Conservation Commission), Dr. Lamberth Kanga (Florida A&M University), and Dr. Muhammad Haseeb (Florida A&M University). I am thankful to all my colleagues and lab mates at the University of Florida who reviewed this manuscript and offered valuable comments and suggestions. I am equally thankful to the USDA for providing funding to this study through the Hydrilla Integrated Pest Management Risk Avoidance and Mitigation Project (IPM RAMP) grant 2010-02825 and the National Institute of Food and Agriculture Crop Protection and Pest Management (NIFA CPPM) grant 2014-70006-22517. -

Testing Agricultural Impacts on Breeding Ground Food Resources As a Driver of Cuckoo Population Decline
Testing agricultural impacts on breeding ground food resources as a driver of cuckoo population decline Submitted by Lowell John Mills to the University of Exeter as a thesis for the degree of Doctor of Philosophy in Biological Sciences, March 2019 This thesis is available for Library use on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. I certify that all material in this thesis which is not my own work has been identified and that no material has previously been submitted and approved for the award of a degree by this or any other university. 1 2 Image: Charles Tyler “The first picture of you, The first picture of summer, Seeing the flowers scream their joy.” - The Lotus Eaters (1983) 3 4 Abstract The common cuckoo Cuculus canorus has undergone a striking divergence in population trend between UK habitats since the 1980s. The breeding population in Scotland – in largely semi-natural open habitat – shows significant increase whereas there has been a significant decline in England. Here breeding numbers have remained stable or increased in semi-natural habitats, while woodland and farmland populations have plummeted. As a brood parasitic bird with a long-distance annual migration, the cuckoo has a unique network of relationships to songbird „hosts‟, prey and habitat; and a disconnection between adult and nestling ecology due to lack of parental care. This thesis investigated the role of breeding ground land-use factors in driving cuckoo population decline. In the first chapter information was synthesised from the literature on potential threats and environmental impacts facing cuckoo populations, which also highlighted knowledge gaps and a basis for hypotheses in later chapters. -

Long‐Term Changes in the Abundance of Flying Insects
CORE Metadata, citation and similar papers at core.ac.uk Provided by Rothamsted Repository Insect Conservation and Diversity (2009) 2, 251–260 doi: 10.1111/j.1752-4598.2009.00062.x Long-term changes in the abundance of flying insects CHRIS R. SHORTALL, ALISON MOORE, EMMA SMITH, MIKE J. HALL, IAN P. WOIWOD and RICHARD HARRINGTON Plant and Invertebrate Ecology Department, Rothamsted Research, Harpenden, Hertfordshire, UK Abstract. 1. For the first time, long-term changes in total aerial insect biomass have been estimated for a wide area of Southern Britain. 2. Various indices of biomass were created for standardised samples from four of the Rothamsted Insect Survey 12.2 m tall suction traps for the 30 years from 1973 to 2002. 3. There was a significant decline in total biomass at Hereford but not at three other sites: Rothamsted, Starcross and Wye. 4. For the Hereford samples, many insects were identified at least to order level, some to family or species level. These samples were then used to investigate the taxa involved in the decline in biomass at Hereford. 5. The Hereford samples were dominated by large Diptera, particularly Dilophus febrilis, which showed a significant decline in abundance. 6. Changes in agricultural practice that could have contributed to the observed declines are discussed, as are potential implications for farmland birds, with sugges- tions for further work to investigate both cause and effect. Key words. Biodiversity, biomass, Diptera, long-term monitoring, suction trap. Introduction undergone well-documented declines in recent years. These declines coincided with a period of agricultural intensification There is widespread concern over biodiversity extinction rates (Buckwell & Armstrong-Brown, 2004; Buckingham et al., 2006), and their impact on the human species (Pimm et al., 1995). -

Chironomidae Hirschkopf
Literatur Chironomidae Gesäuse U.A. zur Bestimmung und Ermittlung der Autökologie herangezogene Literatur: Albu, P. (1972): Două specii de Chironomide noi pentru ştiinţă în masivul Retezat.- St. şi Cerc. Biol., Seria Zoologie, 24: 15-20. Andersen, T.; Mendes, H.F. (2002): Neotropical and Mexican Mesosmittia Brundin, with the description of four new species (Insecta, Diptera, Chironomidae).- Spixiana, 25(2): 141-155. Andersen, T.; Sæther, O.A. (1993): Lerheimia, a new genus of Orthocladiinae from Africa (Diptera: Chironomidae).- Spixiana, 16: 105-112. Andersen, T.; Sæther, O.A.; Mendes, H.F. (2010): Neotropical Allocladius Kieffer, 1913 and Pseudosmittia Edwards, 1932 (Diptera: Chironomidae).- Zootaxa, 2472: 1-77. Baranov, V.A. (2011): New and rare species of Orthocladiinae (Diptera, Chironomidae) from the Crimea, Ukraine.- Vestnik zoologii, 45(5): 405-410. Boggero, A.; Zaupa, S.; Rossaro, B. (2014): Pseudosmittia fabioi sp. n., a new species from Sardinia (Diptera: Chironomidae, Orthocladiinae).- Journal of Entomological and Acarological Research, [S.l.],46(1): 1-5. Brundin, L. (1947): Zur Kenntnis der schwedischen Chironomiden.- Arkiv för Zoologi, 39 A(3): 1- 95. Brundin, L. (1956): Zur Systematik der Orthocladiinae (Dipt. Chironomidae).- Rep. Inst. Freshwat. Drottningholm 37: 5-185. Casas, J.J.; Laville, H. (1990): Micropsectra seguyi, n. sp. du groupe attenuata Reiss (Diptera: Chironomidae) de la Sierra Nevada (Espagne).- Annls Soc. ent. Fr. (N.S.), 26(3): 421-425. Caspers, N. (1983): Chironomiden-Emergenz zweier Lunzer Bäche, 1972.- Arch. Hydrobiol. Suppl. 65: 484-549. Caspers, N. (1987): Chaetocladius insolitus sp. n. (Diptera: Chironomidae) from Lunz, Austria. In: Saether, O.A. (Ed.): A conspectus of contemporary studies in Chironomidae (Diptera). -

Spatial and Temporal Distribution of Aquatic Insects in the Dicle (Tigris) River Basin, Turkey, with New Records
Turkish Journal of Zoology Turk J Zool (2017) 41: 102-112 http://journals.tubitak.gov.tr/zoology/ © TÜBİTAK Research Article doi:10.3906/zoo-1512-56 Spatial and temporal distribution of aquatic insects in the Dicle (Tigris) River Basin, Turkey, with new records Fatma ÇETİNKAYA, Aysel BEKLEYEN* Department of Biology, Faculty of Science, Dicle University, Diyarbakır, Turkey Received: 21.12.2015 Accepted/Published Online: 01.06.2016 Final Version: 25.01.2017 Abstract: We investigated insects of the Dicle (Tigris) River Basin in terms of their composition and spatiotemporal distribution. Larvae, pupae, pupal exuviae, and nymphs of insects were obtained from samples collected by a plankton net monthly during a 1-year period in 2008 and 2009 at seven different sites of the Dicle (Tigris) River Basin. A total of 35 taxa from the orders Trichoptera (1 taxon), Ephemeroptera (3 taxa), and Diptera (31 taxa) were identified. Chironomidae (Diptera) was the most diverse group and was represented by three major subfamilies, namely Tanypodinae (2 taxa), Orthocladiinae (19 taxa), and Chironominae (7 taxa). Among these species, Nanocladius (Nanocladius) spiniplenus Saether, 1977 is a new record for Turkey as well as for western Asia. In addition, the Psychomyia larvae found for the first time in the Dicle (Tigris) River Basin (Turkey) were described. Both taxa have been illustrated to warrant validation. Taxa number varied spatially from 6 to 14 and temporally from 2 to 12 during the sampling period. Along the river, Cricotopus bicinctus and Orthocladius (S.) holsatus were the most common taxa. Key words: Diptera, Ephemeroptera, Trichoptera, Insecta, Dicle (Tigris) River 1. -
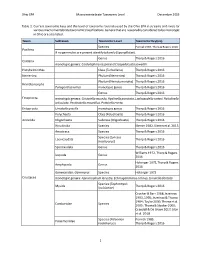
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 1 Table 1. Current Taxonomic Keys and the Level of Taxonomy Routinely U
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Table 1. Current taxonomic keys and the level of taxonomy routinely used by the Ohio EPA in streams and rivers for various macroinvertebrate taxonomic classifications. Genera that are reasonably considered to be monotypic in Ohio are also listed. Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Species Pennak 1989, Thorp & Rogers 2016 Porifera If no gemmules are present identify to family (Spongillidae). Genus Thorp & Rogers 2016 Cnidaria monotypic genera: Cordylophora caspia and Craspedacusta sowerbii Platyhelminthes Class (Turbellaria) Thorp & Rogers 2016 Nemertea Phylum (Nemertea) Thorp & Rogers 2016 Phylum (Nematomorpha) Thorp & Rogers 2016 Nematomorpha Paragordius varius monotypic genus Thorp & Rogers 2016 Genus Thorp & Rogers 2016 Ectoprocta monotypic genera: Cristatella mucedo, Hyalinella punctata, Lophopodella carteri, Paludicella articulata, Pectinatella magnifica, Pottsiella erecta Entoprocta Urnatella gracilis monotypic genus Thorp & Rogers 2016 Polychaeta Class (Polychaeta) Thorp & Rogers 2016 Annelida Oligochaeta Subclass (Oligochaeta) Thorp & Rogers 2016 Hirudinida Species Klemm 1982, Klemm et al. 2015 Anostraca Species Thorp & Rogers 2016 Species (Lynceus Laevicaudata Thorp & Rogers 2016 brachyurus) Spinicaudata Genus Thorp & Rogers 2016 Williams 1972, Thorp & Rogers Isopoda Genus 2016 Holsinger 1972, Thorp & Rogers Amphipoda Genus 2016 Gammaridae: Gammarus Species Holsinger 1972 Crustacea monotypic genera: Apocorophium lacustre, Echinogammarus ischnus, Synurella dentata Species (Taphromysis Mysida Thorp & Rogers 2016 louisianae) Crocker & Barr 1968; Jezerinac 1993, 1995; Jezerinac & Thoma 1984; Taylor 2000; Thoma et al. Cambaridae Species 2005; Thoma & Stocker 2009; Crandall & De Grave 2017; Glon et al. 2018 Species (Palaemon Pennak 1989, Palaemonidae kadiakensis) Thorp & Rogers 2016 1 Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Informal grouping of the Arachnida Hydrachnidia Smith 2001 water mites Genus Morse et al. -

Checklist of the Family Chironomidae (Diptera) of Finland
A peer-reviewed open-access journal ZooKeys 441: 63–90 (2014)Checklist of the family Chironomidae (Diptera) of Finland 63 doi: 10.3897/zookeys.441.7461 CHECKLIST www.zookeys.org Launched to accelerate biodiversity research Checklist of the family Chironomidae (Diptera) of Finland Lauri Paasivirta1 1 Ruuhikoskenkatu 17 B 5, FI-24240 Salo, Finland Corresponding author: Lauri Paasivirta ([email protected]) Academic editor: J. Kahanpää | Received 10 March 2014 | Accepted 26 August 2014 | Published 19 September 2014 http://zoobank.org/F3343ED1-AE2C-43B4-9BA1-029B5EC32763 Citation: Paasivirta L (2014) Checklist of the family Chironomidae (Diptera) of Finland. In: Kahanpää J, Salmela J (Eds) Checklist of the Diptera of Finland. ZooKeys 441: 63–90. doi: 10.3897/zookeys.441.7461 Abstract A checklist of the family Chironomidae (Diptera) recorded from Finland is presented. Keywords Finland, Chironomidae, species list, biodiversity, faunistics Introduction There are supposedly at least 15 000 species of chironomid midges in the world (Armitage et al. 1995, but see Pape et al. 2011) making it the largest family among the aquatic insects. The European chironomid fauna consists of 1262 species (Sæther and Spies 2013). In Finland, 780 species can be found, of which 37 are still undescribed (Paasivirta 2012). The species checklist written by B. Lindeberg on 23.10.1979 (Hackman 1980) included 409 chironomid species. Twenty of those species have been removed from the checklist due to various reasons. The total number of species increased in the 1980s to 570, mainly due to the identification work by me and J. Tuiskunen (Bergman and Jansson 1983, Tuiskunen and Lindeberg 1986). -

A Synopsis of the Biology and Life History of Ruffe
J. Great Lakes Res. 24(2): 170-1 85 Internat. Assoc. Great Lakes Res., 1998 A Synopsis of the Biology and Life History of Ruffe Derek H. Ogle* Northland College Mathematics Department Ashland, Wisconsin 54806 ABSTRACT. The ruffe (Gymnocephalus cernuus), a Percid native to Europe and Asia, has recently been introduced in North America and new areas of Europe. A synopsis of the biology and life history of ruffe suggests a great deal of variability exists in these traits. Morphological characters vary across large geographical scales, within certain water bodies, and between sexes. Ruffe can tolerate a wide variety of conditions including fresh and brackish waters, lacustrine and lotic systems, depths of 0.25 to 85 m, montane and submontane areas, and oligotrophic to eutrophic waters. Age and size at maturity dif- fer according to temperature and levels of mortality. Ruffe spawn on a variety ofsubstrates, for extended periods of time. In some populations, individual ruffe may spawn more than once per year. Growth of ruffe is affected by sex, morphotype, water type, intraspecific density, and food supply. Ruffe feed on a wide variety of foods, although adult ruffe feed predominantly on chironomid larvae. Interactions (i.e., competition and predation) with other species appear to vary considerably between system. INDEX WORDS: Ruffe, review, taxonomy, reproduction, diet, parasite, predation. INTRODUCTION DISTRIBUTION This is a review of the existing literature on Ruffe are native to all of Europe except for along ruffe, providing a synopsis of its biology and life the Mediterranean Sea, western France, Spain, Por- history. A review of the existing literature is tugal, Norway, northern Finland, Ireland, and Scot- needed at this time because the ruffe, which is na- land (Collette and Banarescu 1977, Lelek 1987).