Anticancer DNA Intercalators Cause P53-Dependent Mitochondrial DNA Nucleoid Re-Modelling
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

(NAP) and Illustration of DNA Flexure Angles at Single Molecule Resolution Debayan Purkait†A, Debolina Bandyopadhyay†A, and Padmaja P
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.11.293639; this version posted September 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Vital Insights into Prokaryotic Genome Compaction by Nucleoid-Associated Protein (NAP) and Illustration of DNA Flexure Angles at Single Molecule Resolution Debayan Purkait†a, Debolina Bandyopadhyay†a, and Padmaja P. Mishra*a aChemical Sciences Division, Saha Institute of Nuclear Physics, India aHomi Bhaba National Institute (HBNI), India †Both the authors have contributed equally. * Corresponding author Abstract Integration Host Factor (IHF) is a heterodimeric site-specific nucleoid-associated protein (NAP) well known for its DNA bending ability. The binding is mediated through the narrow minor grooves of the consensus sequence, involving van der-Waals interaction and hydrogen bonding. Although the DNA bend state of IHF has been captured by both X-ray Crystallography and Atomic Force Microscopy (AFM), the range of flexibility and degree of heterogeneity in terms of quantitative analysis of the nucleoprotein complex has largely remained unexplored. Here we have monitored and compared the trajectories of the conformational dynamics of a dsDNA upon binding of wild-type (wt) and single-chain (sc) IHF at millisecond resolution through single-molecule FRET (smFRET). Our findings reveal that the nucleoprotein complex exists in a ‘Slacked-Dynamic’ state throughout the observation window where many of them have switched between multiple ‘Wobbling States’ in the course of attainment of packaged form. A range of DNA ‘Flexure Angles’ has been calculated that give us vital insights regarding the nucleoid organization and transcriptional regulation in prokaryotes. -

Cell Cycle Regulation by the Bacterial Nucleoid
Available online at www.sciencedirect.com ScienceDirect Cell cycle regulation by the bacterial nucleoid David William Adams, Ling Juan Wu and Jeff Errington Division site selection presents a fundamental challenge to all terminate in the terminus region (Ter; 1808). Once organisms. Bacterial cells are small and the chromosome chromosome replication and segregation are complete (nucleoid) often fills most of the cell volume. Thus, in order to the cell is ready to divide. In Bacteria this normally occurs maximise fitness and avoid damaging the genetic material, cell by binary fission and in almost all species this is initiated division must be tightly co-ordinated with chromosome by the assembly of the tubulin homologue FtsZ into a replication and segregation. To achieve this, bacteria employ a ring-like structure (‘Z-ring’) at the nascent division site number of different mechanisms to regulate division site (Figure 1) [1]. The Z-ring then functions as a dynamic selection. One such mechanism, termed nucleoid occlusion, platform for assembly of the division machinery [2,3]. Its allows the nucleoid to protect itself by acting as a template for central role in division also allows FtsZ to serve as a nucleoid occlusion factors, which prevent Z-ring assembly over regulatory hub for the majority of regulatory proteins the DNA. These factors are sequence-specific DNA-binding identified to date [2,4]. Nevertheless, the precise ultra- proteins that exploit the precise organisation of the nucleoid, structure of the Z-ring and whether or not it plays a direct allowing them to act as both spatial and temporal regulators of role in force-generation during division remains contro- bacterial cell division. -

Catalytic Domain of Plasmid Pad1 Relaxase Trax Defines a Group Of
Catalytic domain of plasmid pAD1 relaxase TraX defines a group of relaxases related to restriction endonucleases María Victoria Franciaa,1, Don B. Clewellb,c, Fernando de la Cruzd, and Gabriel Moncaliánd aServicio de Microbiología, Hospital Universitario Marqués de Valdecilla e Instituto de Formación e Investigación Marqués de Valdecilla, Santander 39008, Spain; bDepartment of Biologic and Materials Sciences, University of Michigan School of Dentistry, Ann Arbor, MI 48109; cDepartment of Microbiology and Immunology, University of Michigan Medical School, Ann Arbor, MI 48109; and dDepartamento de Biología Molecular e Instituto de Biomedicina y Biotecnología de Cantabria, Universidad de Cantabria–Consejo Superior de Investigaciones Científicas–Sociedad para el Desarrollo Regional de Cantabria, Santander 39011, Spain Edited by Roy Curtiss III, Arizona State University, Tempe, AZ, and approved July 9, 2013 (received for review May 30, 2013) Plasmid pAD1 is a 60-kb conjugative element commonly found in known relaxases show two characteristic sequence motifs, motif I clinical isolates of Enterococcus faecalis. The relaxase TraX and the containing the catalytic Tyr residue (which covalently attaches to primary origin of transfer oriT2 are located close to each other and the 5′ end of the cleaved DNA) and motif III with the His-triad have been shown to be essential for conjugation. The oriT2 site essential for relaxase activity (facilitating the cleavage reaction contains a large inverted repeat (where the nic site is located) by activation of the catalytic Tyr). This His-triad has been used as adjacent to a series of short direct repeats. TraX does not show a relaxase diagnostic signature (12). fi any of the typical relaxase sequence motifs but is the prototype of Plasmid pAD1 relaxase, TraX, was identi ed (9) and shown to oriT2 a unique family of relaxases (MOB ). -

Relaxation Complexes of Plasmids Colel and Cole2:Unique Site of The
Proc. Nat. Acad. Sci. USA Vol. 71, No. 10, pp. 3854-3857, October 1974 Relaxation Complexes of Plasmids ColEl and ColE2: Unique Site of the Nick in the Open Circular DNA of the Relaxed Complexes (Escherichia coli/supercoiled DNA/endonuclease/restriction enzyme/DNA replication) MICHAEL A. LOVETT, DONALD G. GUINEY, AND DONALD R. HELINSKI Department of Biology, University of California, San Diego, La Jolla, Calif. 92037 Communicated by Stanley L. Miller, June 26, 1974 ABSTRACT The product of the induced relaxation of volved in the production of a single-strand cleavage in the supercoiled DNA-protein relaxation complexes of colicino- initiation of replication and/or conjugal transfer of plasmid genic factors El (ColEl) and E2 (CoIE2) is an open circular DNA molecule with a strand-specific nick. Cleavage of the DNA. An involvement in such processes would most likely open circular DNA of each relaxed complex with the EcoRI require that the relaxation event take place at a unique site restriction endonuclease demonstrates that the single- in the DNA molecule. In this report, evidence will be pre- strand break is at a unique position. The site of the single- sented from studies using the EcoRI restriction endonuclease strand break in the relaxed ColEl complex is approximately the same distance from the EcoRI cleavage site as the that the strand-specific relaxation event takes place at a origin of ColEl DNA replication. unique site on both the ColEl and CoIE2 plasmid molecules. The location of this site, at least in the case of the ColEl Many of extrachromosomal circular DNA elements (plas- plasmid, is approximately the same distance from the single mids) of Escherichia coli, differing in molecular weight, num- EcoRI site as the origin of replication for this plasmid. -
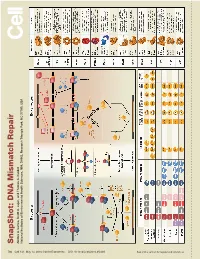
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

Effects of Abasic Sites and DNA Single-Strand Breaks on Prokaryotic RNA Polymerases (Apurinic/Apyrimidinic Sites/Transcription/DNA Damage) WEI ZHOU* and PAUL W
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 6601-6605, July 1993 Biochemistry Effects of abasic sites and DNA single-strand breaks on prokaryotic RNA polymerases (apurinic/apyrimidinic sites/transcription/DNA damage) WEI ZHOU* AND PAUL W. DOETSCH*tt Departments of *Biochemistry and tRadiation Oncology and tDivision of Cancer Biology, Emory University School of Medicine, Atlanta, GA 30322 Communicated by Philip C. Hanawalt, April 19, 1993 ABSTRACT Abasic sites are thought to be the most fre- effects of such damage on both transcription initiation and quently occurring cellular DNA damage and are generated elongation processes (19, 20). We wished to determine the spontaneously or as the result of chemical or radiation damage effects of AP sites on the transcription elongation process by to DNA. In contrast to the wealth of information that exists on using a defined system that would provide direct information the effects of abasic sites on DNA polymerases, very little is regarding the ability of RNA polymerases to bypass or be known about how these lesions interact with RNA polymerases. blocked by AP sites and, if bypass was observed, to deter- An in vitro transcription system was used to determine the mine the nature of the base insertion opposite such lesions. effects of abasic sites and single-strand breaks on transcrip- For these studies, we constructed a DNA template containing tional elongation. DNA templates were constructed containing a single AP site placed at a unique location downstream from single abasic sites or nicks placed at unique locations down- either the SP6 or tac promoter and carried out in vitro stream from two different promoters and were transcribed by transcription experiments with SP6 and E. -

DNA Structure at the Plasmid Origin-Of-Transfer Indicates Its
www.nature.com/scientificreports OPEN DNA structure at the plasmid origin-of-transfer indicates its potential transfer range Received: 12 May 2016 Jan Zrimec1,2,3 & Aleš Lapanje1,4,5 Accepted: 10 January 2018 Horizontal gene transfer via plasmid conjugation enables antimicrobial resistance (AMR) to spread Published: xx xx xxxx among bacteria and is a major health concern. The range of potential transfer hosts of a particular conjugative plasmid is characterised by its mobility (MOB) group, which is currently determined based on the amino acid sequence of the plasmid-encoded relaxase. To facilitate prediction of plasmid MOB groups, we have developed a bioinformatic procedure based on analysis of the origin-of-transfer (oriT), a merely 230 bp long non-coding plasmid DNA region that is the enzymatic substrate for the relaxase. By computationally interpreting conformational and physicochemical properties of the oriT region, which facilitate relaxase-oriT recognition and initiation of nicking, MOB groups can be resolved with over 99% accuracy. We have shown that oriT structural properties are highly conserved and can be used to discriminate among MOB groups more efciently than the oriT nucleotide sequence. The procedure for prediction of MOB groups and potential transfer range of plasmids was implemented using published data and is available at http://dnatools.eu/MOB/plasmid.html. Antimicrobial resistance (AMR) is a pressing global issue, as it diminishes the activity of 29 antibiotics and conse- quently leads to over 25,000 deaths each year in Europe alone1,2. Te development of AMR in microbial commu- nities is facilitated by horizontal gene transfer (HGT) of conjugative elements (including plasmids and integrative elements)3 carrying antibiotic resistance genes along with virulence genes4,5. -

Enzymatic Completion of Mammalian Lagging-Strand DNA Replication JOHN J
Proc. Natl. Acad. Sci. USA Vol. 91, pp. 9803-9807, October 1994 Biochemistry Enzymatic completion of mammalian lagging-strand DNA replication JOHN J. TuRCHI*, LIN HUANGt, RICHARD S. MURANTEt, YONG KIMt, AND ROBERT A. BAMBARAtt *Department of Biochemistry and Molecular Biology, Wright State University, Dayton, OH 45435; and tDepartment of Biochemistry, University of Rochester, Rochester, NY 14642 Communicated by Fred Sherman, June 8, 1994 (receivedfor review April 28, 1994) ABSTRACT Using purified proteins from calf and a syn- in DNA replication and transcription (21), but no specific thetic substrate, we have reconstituted the enzymatic reactions roles have been established for either class. required for mammalian Okazaki fragment processing in vitro. We had shown (22) that the calf 5'-to-3' exonuclease could The required reactions are removal of initiator RNA, synthesis work with calf DNA polymerases a, 8, or e and DNA ligase fromanupstre fragment to generate a nick, and thenligation. I to join two DNA primers annealed to a template with a With our substrate, RNase H type I (RNase HI) makes a single four-base gap. In addition, we have demonstrated that 5'- cut in the initiator RNA, one nucleotide 5' of the RNA-DNA to-3' exonuclease activity is stimulated by synthesis from an junction. The double strandspecific 5' to3' exonuclease removes upstream primer and that the stimulation is the result of the the remaining monoribonuceotide. After dissociation ofcleaved formation of a nick between the two primers, which is a RNA, synthesis by DNA polymerase generates a nick, which is preferred substrate for the exonuclease (17). These results then sealed by DNA igase I. -

Can a Nick Promote Partial Genome Re-Replication?
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.14.381277; this version posted November 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Can a nick promote partial genome re-replication? Erik Johansson1,2,* and John F.X Diffley1,* 1 Chromosome Replication Laboratory The Francis Crick Institute, 1 Midland Road, London, NW1 1AT 2 Dept. of Medical Biochemistry and Biophysics, Umeå University, 90187 Umeå, Sweden *Corresponding Authors [email protected] Tel: +46 (0) 90 786 6638 [email protected] Tel: +44 (0) 203 796 1833 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.14.381277; this version posted November 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Single-stranded DNA breaks, including simple nicks, are amongst the most common forms of DNA damage in cells. They can be readily repaired by ligation; however, if a nick occurs just ahead of an approaching replisome, the outcome is a ‘collapsed’ replication fork in which the nick is converted into a single-ended double-strand DNA break. Attention has largely focused on the processes by which this broken end is used to prime replication restart. -

Direct Observation of Topoisomerase IA Gate Dynamics Maria Mills1, Yuk-Ching Tse-Dinh2,3, & Keir C
bioRxiv preprint doi: https://doi.org/10.1101/356659; this version posted June 27, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. Direct Observation of Topoisomerase IA Gate Dynamics Maria Mills1, Yuk-Ching Tse-Dinh2,3, & Keir C. Neuman1* 1 Laboratory of Single Molecule Biophysics, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, Maryland 20892, USA 2 Biomolecular Sciences Institute, Florida International University, Miami, Florida 33199, USA 3 Department of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, USA * Corresponding author. Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/356659; this version posted June 27, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. Abstract Type IA topoisomerases cleave single-stranded DNA and relieve negative supercoils in discrete steps corresponding to the passage of the intact DNA strand through the cleaved strand. Although it is assumed type IA topoisomerases accomplish this strand passage via a protein-mediated DNA gate, opening of this gate has never been observed. We developed a single-molecule assay to directly measure gate opening of the E. coli type IA topoisomerases I and III. -

The Devil Is in the Details for DNA Mismatch Repair COMMENTARY
COMMENTARY The Devil is in the details for DNA mismatch repair COMMENTARY Peggy Hsieha,1 and Yongliang Zhanga Ensuring high-fidelity DNA replication is essential a strand-specific signal for excision can be far apart) (7–9). for maintaining genome stability in organisms from MMR in eukaryotes features two families of MMR pro- Escherichia coli to humans. This task requires an intricate teins, heterodimeric homologs of bacterial MutS (MSH) network of cellular components that carries out replica- or MutL (MLH). Early models of E. coli MMR invoke tion and postreplication DNA repair (1). In eukaryotes, MMR protein-mediated DNA looping between a mis- replication is carried out by Pol e and Pol δ that synthesize match and a DNA-methylation excision signal or ATP- the leading and lagging strands, respectively, using short driven MutS translocation along the DNA. The nucleotide RNA–DNA primers synthesized by Pol α. Although repli- switch model posits the existence of multiple diffusing cation gives rise to a low spontaneous mutation rate of MSH–MLH clamps (8). Other studies suggest that a single − ∼10 10 mutations per base pair per generation, errors in MSH clamp at a mismatch can load multiple MLH clamps the nucleotide incorporation step occur about once every to license excision (1, 9). MutSα (MSH2-MSH6) and MutSβ + 104 to 105 insertions on average, although the frequency (MSH2-MSH3) are related to the AAA family of ATPases varies considerably depending on a number of parame- and carry out mismatch recognition (10, 11). MutL homo- ters including the particular mismatch, chromosomal con- logs MutLα (yeast ScMlh1-Pms1 and human HsMLH1- text, and the DNA polymerase. -

Nickseq for Genome-Wide Strand-Specific Identification of DNA Single- Strand Break Sites with Single Nucleotide Resolution
bioRxiv preprint doi: https://doi.org/10.1101/867937; this version posted December 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. NickSeq for genome-wide strand-specific identification of DNA single- strand break sites with single nucleotide resolution Joshua J. Elacqua1,2,*, Navpreet Ranu1,2,4,*, Sarah E. DiIorio1,2,5 and Paul C. Blainey1,2,3,+ ABSTRACT INTRODUCTION DNA single-strand breaks (SSBs), or ‘nicks’, are Cells continuously accrue DNA damage due to the most common form of DNA damage. Nicks exposure to environmental stressors. The single- occur at rates of tens of thousands per cell per strand break (SSB), or ‘nick’, is estimated to day, and result from many sources including occur at rates of tens of thousands per cell per oxidative stress and endogenous enzyme day, making it the most frequent type of damage.1 activities. Accumulation of nicks, due to high rates Reactive oxygen species such as hydrogen of occurrence or defects in repair enzymes, has peroxide-derived free radicals can nick DNA been implicated in multiple diseases. However, either directly by reacting with its sugar- improved methods for nick analysis are needed phosphate backbone or indirectly by altering to learn how their locations and number affect nucleobases which are then subject to repair cells, disease progression, and health outcomes. pathways entailing nicked intermediates.1,2 In addition to natural processes including DNA Endogenous processes including DNA repair, leading genome-editing technologies rely replication and topoisomerase activity also on nuclease activity, including nick generation, at generate nicks.1,3 target sites.