DNA Structure at the Plasmid Origin-Of-Transfer Indicates Its
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(NAP) and Illustration of DNA Flexure Angles at Single Molecule Resolution Debayan Purkait†A, Debolina Bandyopadhyay†A, and Padmaja P
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.11.293639; this version posted September 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Vital Insights into Prokaryotic Genome Compaction by Nucleoid-Associated Protein (NAP) and Illustration of DNA Flexure Angles at Single Molecule Resolution Debayan Purkait†a, Debolina Bandyopadhyay†a, and Padmaja P. Mishra*a aChemical Sciences Division, Saha Institute of Nuclear Physics, India aHomi Bhaba National Institute (HBNI), India †Both the authors have contributed equally. * Corresponding author Abstract Integration Host Factor (IHF) is a heterodimeric site-specific nucleoid-associated protein (NAP) well known for its DNA bending ability. The binding is mediated through the narrow minor grooves of the consensus sequence, involving van der-Waals interaction and hydrogen bonding. Although the DNA bend state of IHF has been captured by both X-ray Crystallography and Atomic Force Microscopy (AFM), the range of flexibility and degree of heterogeneity in terms of quantitative analysis of the nucleoprotein complex has largely remained unexplored. Here we have monitored and compared the trajectories of the conformational dynamics of a dsDNA upon binding of wild-type (wt) and single-chain (sc) IHF at millisecond resolution through single-molecule FRET (smFRET). Our findings reveal that the nucleoprotein complex exists in a ‘Slacked-Dynamic’ state throughout the observation window where many of them have switched between multiple ‘Wobbling States’ in the course of attainment of packaged form. A range of DNA ‘Flexure Angles’ has been calculated that give us vital insights regarding the nucleoid organization and transcriptional regulation in prokaryotes. -

Cell Cycle Regulation by the Bacterial Nucleoid
Available online at www.sciencedirect.com ScienceDirect Cell cycle regulation by the bacterial nucleoid David William Adams, Ling Juan Wu and Jeff Errington Division site selection presents a fundamental challenge to all terminate in the terminus region (Ter; 1808). Once organisms. Bacterial cells are small and the chromosome chromosome replication and segregation are complete (nucleoid) often fills most of the cell volume. Thus, in order to the cell is ready to divide. In Bacteria this normally occurs maximise fitness and avoid damaging the genetic material, cell by binary fission and in almost all species this is initiated division must be tightly co-ordinated with chromosome by the assembly of the tubulin homologue FtsZ into a replication and segregation. To achieve this, bacteria employ a ring-like structure (‘Z-ring’) at the nascent division site number of different mechanisms to regulate division site (Figure 1) [1]. The Z-ring then functions as a dynamic selection. One such mechanism, termed nucleoid occlusion, platform for assembly of the division machinery [2,3]. Its allows the nucleoid to protect itself by acting as a template for central role in division also allows FtsZ to serve as a nucleoid occlusion factors, which prevent Z-ring assembly over regulatory hub for the majority of regulatory proteins the DNA. These factors are sequence-specific DNA-binding identified to date [2,4]. Nevertheless, the precise ultra- proteins that exploit the precise organisation of the nucleoid, structure of the Z-ring and whether or not it plays a direct allowing them to act as both spatial and temporal regulators of role in force-generation during division remains contro- bacterial cell division. -

Catalytic Domain of Plasmid Pad1 Relaxase Trax Defines a Group Of
Catalytic domain of plasmid pAD1 relaxase TraX defines a group of relaxases related to restriction endonucleases María Victoria Franciaa,1, Don B. Clewellb,c, Fernando de la Cruzd, and Gabriel Moncaliánd aServicio de Microbiología, Hospital Universitario Marqués de Valdecilla e Instituto de Formación e Investigación Marqués de Valdecilla, Santander 39008, Spain; bDepartment of Biologic and Materials Sciences, University of Michigan School of Dentistry, Ann Arbor, MI 48109; cDepartment of Microbiology and Immunology, University of Michigan Medical School, Ann Arbor, MI 48109; and dDepartamento de Biología Molecular e Instituto de Biomedicina y Biotecnología de Cantabria, Universidad de Cantabria–Consejo Superior de Investigaciones Científicas–Sociedad para el Desarrollo Regional de Cantabria, Santander 39011, Spain Edited by Roy Curtiss III, Arizona State University, Tempe, AZ, and approved July 9, 2013 (received for review May 30, 2013) Plasmid pAD1 is a 60-kb conjugative element commonly found in known relaxases show two characteristic sequence motifs, motif I clinical isolates of Enterococcus faecalis. The relaxase TraX and the containing the catalytic Tyr residue (which covalently attaches to primary origin of transfer oriT2 are located close to each other and the 5′ end of the cleaved DNA) and motif III with the His-triad have been shown to be essential for conjugation. The oriT2 site essential for relaxase activity (facilitating the cleavage reaction contains a large inverted repeat (where the nic site is located) by activation of the catalytic Tyr). This His-triad has been used as adjacent to a series of short direct repeats. TraX does not show a relaxase diagnostic signature (12). fi any of the typical relaxase sequence motifs but is the prototype of Plasmid pAD1 relaxase, TraX, was identi ed (9) and shown to oriT2 a unique family of relaxases (MOB ). -

Relaxation Complexes of Plasmids Colel and Cole2:Unique Site of The
Proc. Nat. Acad. Sci. USA Vol. 71, No. 10, pp. 3854-3857, October 1974 Relaxation Complexes of Plasmids ColEl and ColE2: Unique Site of the Nick in the Open Circular DNA of the Relaxed Complexes (Escherichia coli/supercoiled DNA/endonuclease/restriction enzyme/DNA replication) MICHAEL A. LOVETT, DONALD G. GUINEY, AND DONALD R. HELINSKI Department of Biology, University of California, San Diego, La Jolla, Calif. 92037 Communicated by Stanley L. Miller, June 26, 1974 ABSTRACT The product of the induced relaxation of volved in the production of a single-strand cleavage in the supercoiled DNA-protein relaxation complexes of colicino- initiation of replication and/or conjugal transfer of plasmid genic factors El (ColEl) and E2 (CoIE2) is an open circular DNA molecule with a strand-specific nick. Cleavage of the DNA. An involvement in such processes would most likely open circular DNA of each relaxed complex with the EcoRI require that the relaxation event take place at a unique site restriction endonuclease demonstrates that the single- in the DNA molecule. In this report, evidence will be pre- strand break is at a unique position. The site of the single- sented from studies using the EcoRI restriction endonuclease strand break in the relaxed ColEl complex is approximately the same distance from the EcoRI cleavage site as the that the strand-specific relaxation event takes place at a origin of ColEl DNA replication. unique site on both the ColEl and CoIE2 plasmid molecules. The location of this site, at least in the case of the ColEl Many of extrachromosomal circular DNA elements (plas- plasmid, is approximately the same distance from the single mids) of Escherichia coli, differing in molecular weight, num- EcoRI site as the origin of replication for this plasmid. -

The Obscure World of Integrative and Mobilizable Elements Gérard Guédon, Virginie Libante, Charles Coluzzi, Sophie Payot-Lacroix, Nathalie Leblond-Bourget
The obscure world of integrative and mobilizable elements Gérard Guédon, Virginie Libante, Charles Coluzzi, Sophie Payot-Lacroix, Nathalie Leblond-Bourget To cite this version: Gérard Guédon, Virginie Libante, Charles Coluzzi, Sophie Payot-Lacroix, Nathalie Leblond-Bourget. The obscure world of integrative and mobilizable elements: Highly widespread elements that pirate bacterial conjugative systems. Genes, MDPI, 2017, 8 (11), pp.337. 10.3390/genes8110337. hal- 01686871 HAL Id: hal-01686871 https://hal.archives-ouvertes.fr/hal-01686871 Submitted on 26 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License G C A T T A C G G C A T genes Review The Obscure World of Integrative and Mobilizable Elements, Highly Widespread Elements that Pirate Bacterial Conjugative Systems Gérard Guédon *, Virginie Libante, Charles Coluzzi, Sophie Payot and Nathalie Leblond-Bourget * ID DynAMic, Université de Lorraine, INRA, 54506 Vandœuvre-lès-Nancy, France; [email protected] (V.L.); [email protected] (C.C.); [email protected] (S.P.) * Correspondence: [email protected] (G.G.); [email protected] (N.L.-B.); Tel.: +33-037-274-5142 (G.G.); +33-037-274-5146 (N.L.-B.) Received: 12 October 2017; Accepted: 15 November 2017; Published: 22 November 2017 Abstract: Conjugation is a key mechanism of bacterial evolution that involves mobile genetic elements. -

Virus World As an Evolutionary Network of Viruses and Capsidless Selfish Elements
Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements Koonin, E. V., & Dolja, V. V. (2014). Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements. Microbiology and Molecular Biology Reviews, 78(2), 278-303. doi:10.1128/MMBR.00049-13 10.1128/MMBR.00049-13 American Society for Microbiology Version of Record http://cdss.library.oregonstate.edu/sa-termsofuse Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements Eugene V. Koonin,a Valerian V. Doljab National Center for Biotechnology Information, National Library of Medicine, Bethesda, Maryland, USAa; Department of Botany and Plant Pathology and Center for Genome Research and Biocomputing, Oregon State University, Corvallis, Oregon, USAb Downloaded from SUMMARY ..................................................................................................................................................278 INTRODUCTION ............................................................................................................................................278 PREVALENCE OF REPLICATION SYSTEM COMPONENTS COMPARED TO CAPSID PROTEINS AMONG VIRUS HALLMARK GENES.......................279 CLASSIFICATION OF VIRUSES BY REPLICATION-EXPRESSION STRATEGY: TYPICAL VIRUSES AND CAPSIDLESS FORMS ................................279 EVOLUTIONARY RELATIONSHIPS BETWEEN VIRUSES AND CAPSIDLESS VIRUS-LIKE GENETIC ELEMENTS ..............................................280 Capsidless Derivatives of Positive-Strand RNA Viruses....................................................................................................280 -

The Bacterial Conjugation Protein Trwb Resembles Ring Helicases And
letters to nature metabolically labelled with BrdU (10 mM, 4 h), trypsinized, and ®xed with 70% ethanol. 17. Stampfer, M. R. et al. Gradual phenotypic conversion associated with immortalization of cultured Nuclei were isolated and stained with propidium iodide and FITC-conjugated anti-BrdU human mammary epithelial cells. Mol. Biol. Cell 8, 2391±2405 (1997). antibodies (Becton Dickinson, USA), as described7. Flow cytometry was performed on a 18. Karlseder, J., Broccoli, D., Dai, Y., Hardy, S. & de Lange, T. p53- and ATM-dependent apoptosis FACS Sorter (Becton Dickinson). All analysed events were gated to remove debris and induced by telomeres lacking TRF2. Science 283, 1321±1325 (1999). aggregates. 19. Artandi, S. E. et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406, 641±645 (2000). Cell death assays 20. Chin, L. et al. p53 De®ciency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97, 527±538 (1999). TUNEL assay for DNA fragmentation was done using an In Situ Cell Death Detection kit 21. Alcorta, D. A. et al. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4A) in replicative (BMB), according to manufacturer's protocol. Alternatively, cells were stained with senescence of normal human ®broblasts. Proc. Natl Acad. Sci. USA 92, 13742±13747 (1996). Annexin-V-FLUOR (BMB) and propidium iodide, and analysed by ¯uorescence 22. Hara, E. et al. Regulation of p16CDKN2 expression and its implications for cell immortalization and microscopy. senescence. Mol. Cell. Biol. 16, 859±867 (1996). 23. Burbano, R. R. et al. -

Sequence-Specific DNA Nicking Endonucleases
BioMol Concepts 2015; 6(4): 253–267 Review Open Access Shuang-yong Xu* Sequence-specific DNA nicking endonucleases DOI 10.1515/bmc-2015-0016 Received May 20, 2015; accepted June 24, 2015 Introduction In this article, I will discuss natural DNA nicking endo- nucleases (NEases or nickases) with 3- to 7-bp specificities, Abstract: A group of small HNH nicking endonucleases e.g. Nt.CviPII (↓CCD, the down arrow indicates the nicked (NEases) was discovered recently from phage or prophage strand as shown) originally found in chlorella virus (1), genomes that nick double-stranded DNA sites ranging engineered NEases such as Nt.BspQI (GCTCTTCN↓), and from 3 to 5 bp in the presence of Mg2+ or Mn2+. The cosN site Nt.BbvCI (CC↓TCAGC) engineered from BspQI and BbvCI of phage HK97 contains a gp74 nicking site AC↑CGC, which restriction endonucleases (REases) (2, 3). The other group is similar to AC↑CGR (R = A/G) of N.φGamma encoded by of DNA NEases contains natural or engineered enzymes Bacillus phage Gamma. A minimal nicking domain of 76 with more than 8-bp target sites, which includes group I amino acid residues from N.φGamma could be fused to intron-encoded homing endonucleases (HEs) (4, 5), other DNA binding partners to generate chimeric NEases engineered nicking variants from LAGLIDAG HEs (6–8), with new specificities. The biological roles of a few small engineered TALE nucleases (TALENs) by fusion of tran- HNH endonucleases (HNHE, gp74 of HK97, gp37 of φSLT, scription activator-like effector (TALE) repeat domain with φ12 HNHE) have been demonstrated in phage and patho- FokI nuclease domain or a MutH nicking variant (9–11), ZF genicity island DNA packaging. -
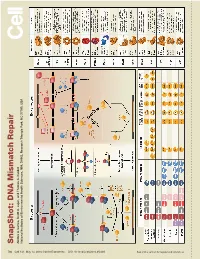
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

Effects of Abasic Sites and DNA Single-Strand Breaks on Prokaryotic RNA Polymerases (Apurinic/Apyrimidinic Sites/Transcription/DNA Damage) WEI ZHOU* and PAUL W
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 6601-6605, July 1993 Biochemistry Effects of abasic sites and DNA single-strand breaks on prokaryotic RNA polymerases (apurinic/apyrimidinic sites/transcription/DNA damage) WEI ZHOU* AND PAUL W. DOETSCH*tt Departments of *Biochemistry and tRadiation Oncology and tDivision of Cancer Biology, Emory University School of Medicine, Atlanta, GA 30322 Communicated by Philip C. Hanawalt, April 19, 1993 ABSTRACT Abasic sites are thought to be the most fre- effects of such damage on both transcription initiation and quently occurring cellular DNA damage and are generated elongation processes (19, 20). We wished to determine the spontaneously or as the result of chemical or radiation damage effects of AP sites on the transcription elongation process by to DNA. In contrast to the wealth of information that exists on using a defined system that would provide direct information the effects of abasic sites on DNA polymerases, very little is regarding the ability of RNA polymerases to bypass or be known about how these lesions interact with RNA polymerases. blocked by AP sites and, if bypass was observed, to deter- An in vitro transcription system was used to determine the mine the nature of the base insertion opposite such lesions. effects of abasic sites and single-strand breaks on transcrip- For these studies, we constructed a DNA template containing tional elongation. DNA templates were constructed containing a single AP site placed at a unique location downstream from single abasic sites or nicks placed at unique locations down- either the SP6 or tac promoter and carried out in vitro stream from two different promoters and were transcribed by transcription experiments with SP6 and E. -

Plasmid Replication-Associated Single-Strand-Specific
12858–12873 Nucleic Acids Research, 2020, Vol. 48, No. 22 Published online 3 December 2020 doi: 10.1093/nar/gkaa1163 Plasmid replication-associated single-strand-specific methyltransferases Alexey Fomenkov 1,*, Zhiyi Sun1, Iain A. Murray 1, Cristian Ruse1, Colleen McClung1, Yoshiharu Yamaichi 2, Elisabeth A. Raleigh 1,* and Richard J. Roberts1,* 1New England Biolabs Inc., 240 County Road, Ipswich, MA, USA and 2Universite´ Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), Gif-sur-Yvette, France Downloaded from https://academic.oup.com/nar/article/48/22/12858/6018438 by guest on 24 September 2021 Received August 06, 2020; Revised November 10, 2020; Editorial Decision November 11, 2020; Accepted November 12, 2020 ABSTRACT RM-associated modification and the diversity of associ- ated functions remains incompletely understood (3). Long- Analysis of genomic DNA from pathogenic strains read, modification-sensitive SMRT sequencing technology of Burkholderia cenocepacia J2315 and Escherichia has facilitated sequencing and assembly of a wide variety coli O104:H4 revealed the presence of two unusual of genomes and also clarified the modification repertoire. MTase genes. Both are plasmid-borne ORFs, carried Detection of the modification status of bases is possible by pBCA072 for B. cenocepacia J2315 and pESBL for for m6A, m4C and oxidized forms of m5C modified bases E. coli O104:H4. Pacific Biosciences SMRT sequenc- (m5hC and 5caC) (4). Recently, high throughput analy- ing was used to investigate DNA methyltransferases sis of 230 diverse bacterial and archaeal methylomes strik- M.BceJIII and M.EcoGIX, using artificial constructs. ingly revealed that almost 50% of organisms harbor Type Mating properties of engineered pESBL derivatives II DNA methyltransferases (MTase) homologs with no ap- were also investigated. -

Structural Basis of a Histidine-DNA Nicking/Joining Mechanism for Gene Transfer and Promiscuous Spread of Antibiotic Resistance
Structural basis of a histidine-DNA nicking/joining mechanism for gene transfer and promiscuous spread of antibiotic resistance Radoslaw Plutaa,b,1,2, D. Roeland Boera,b,1,3, Fabián Lorenzo-Díazc,4, Silvia Russia,b,5, Hansel Gómeza,d, Cris Fernández-Lópezc, Rosa Pérez-Luquea,b, Modesto Orozcoa,d,e, Manuel Espinosac, and Miquel Colla,b,6 aInstitute for Research in Biomedicine, Barcelona Institute of Science and Technology, 08028 Barcelona, Spain; bMolecular Biology Institute of Barcelona, Consejo Superior de Investigaciones Científicas, 08028 Barcelona, Spain; cBiological Research Center, Consejo Superior de Investigaciones Científicas, 28040 Madrid, Spain; dJoint BSC-IRB Research Program in Computational Biology, Institute for Research in Biomedicine, Barcelona Institute of Science and Technology, 08028 Barcelona, Spain; and eDepartment of Biochemistry and Molecular Biology, University of Barcelona, 08028 Barcelona, Spain Edited by Nancy L. Craig, Johns Hopkins University School of Medicine, Baltimore, MD, and approved June 28, 2017 (received for review February 23, 2017) Relaxases are metal-dependent nucleases that break and join DNA the relaxase assembles with other proteins participating in HGT, for the initiation and completion of conjugative bacterial gene namely the coupling protein and the proteins that constitute the transfer. Conjugation is the main process through which antibiotic type IV secretion system (T4SS). To date, relaxase-mediated nu- resistance spreads among bacteria, with multidrug-resistant staph- cleophilic attack at the nic site has been shown to generate a co- ylococci and streptococci infections posing major threats to human valent linkage between a tyrosine residue and the 5′-phosphate health. The MOBV family of relaxases accounts for approximately DNA of the cleaved dinucleotide (8).