Molecular Cloning, Insertional Inactivation And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recruitment of Genes and Enzymes Conferring Resistance to the Nonnatural Toxin Bromoacetate
Recruitment of genes and enzymes conferring resistance to the nonnatural toxin bromoacetate Kevin K. Desai and Brian G. Miller1 Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306-4390 Edited* by Richard Wolfenden, University of North Carolina, Chapel Hill, NC, and approved August 24, 2010 (received for review May 28, 2010) Microbial niches contain toxic chemicals capable of forcing organ- tance of a naïve bacterial population can play a role in combating isms into periods of intense natural selection to afford survival. the toxicity of a nonnatural small-molecule. Revealing the reser- Elucidating the mechanisms by which microbes evade environmen- voir of intrinsic resistance genes that are subject to evolutionary tal threats has direct relevance for understanding and combating recruitment promises to aid our understanding of the processes the rise of antibiotic resistance. In this study we used a toxic small- leading to the emergence of antibiotic resistant pathogens. molecule, bromoacetate, to model the selective pressures imposed We sought to identify the full spectrum of bromoacetate resis- by antibiotics and anthropogenic toxins. We report the results tance mechanisms available to the model bacterium, Escherichia of genetic selection experiments that identify nine genes from coli. The reactivity of bromoacetate is likely to mimic that of elec- Escherichia coli whose overexpression affords survival in the trophilic natural products as well as anthropogenic environmen- presence of a normally lethal concentration of bromoacetate. Eight tal contaminants that microbes may encounter. The clinically of these genes encode putative transporters or transmembrane significant natural antibiotic fosfomycin, and the fungal natural proteins, while one encodes the essential peptidoglycan biosyn- product terreic acid, are electrophilic molecules that both target N thetic enzyme, UDP- -acetylglucosamine enolpyruvoyl transferase an essential nucleophilic cysteine residue in bacteria (8, 9). -

Structural Characterization of Beta Carbonic Anhydrases from Higher Plants
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1998 Structural Characterization of Beta Carbonic Anhydrases From Higher Plants. Michael H. Bracey Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Bracey, Michael H., "Structural Characterization of Beta Carbonic Anhydrases From Higher Plants." (1998). LSU Historical Dissertations and Theses. 6655. https://digitalcommons.lsu.edu/gradschool_disstheses/6655 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type o f computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

Tetranychus Urticae
UvA-DARE (Digital Academic Repository) The role of horizontally transferred genes in the xenobiotic adaptations of the spider mite Tetranychus urticae Wybouw, N.R. Publication date 2015 Document Version Final published version Link to publication Citation for published version (APA): Wybouw, N. R. (2015). The role of horizontally transferred genes in the xenobiotic adaptations of the spider mite Tetranychus urticae. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:03 Oct 2021 Nicky-ch2_Vera-ch1.qxd 18/08/2015 14:33 Page 41 2 A Horizontally Transferred Cyanase Gene in the Spider Mite Tetranychus urticae is Involved in Cyanate Metabolism and is Differentially Expressed Upon Host Plant Change N. Wybouw*, V. Balabanidou*, D.J. Ballhorn, W. -

Enzymatic Study of Cyanide Utilizing Pseudomonas Species Isolated
Journal of Scientific and Innovative Research 2013; 2 (6): 1058-1066 Available online at: www.jsirjournal.com Research Article Enzymatic study of cyanide utilizing Pseudomonas ISSN 2320-4818 species isolated from contaminated soil JSIR 2013; 2(6): 1058-1066 © 2013, All rights reserved Received: 21-10-2013 Dr. Preeti Parmar*, Anjali Soni, Piyush Desai Accepted: 28-12-2013 Abstract Dr. Preeti Parmar Present study deals with understanding the probable metabolic pathway utilized to degrade Department of Biosciences, Veer cyanide present in the bacteria isolated from contaminated soil harboring the loads of industrial Narmad South Gujarat University, effluent mainly rich in cyanide compounds. This was done by performing the enzymatic assay Surat, Gujarat 395007, India of the enzymes used in the pathway. Present studies were conducted on cyanide utilizing Pseudomonas species isolated from previous work of isolation and characterization. It is Anjali Soni supposed that the growing Pseudomonas species would be able to evolve a mechanism to utilize Department of Biotechnology, Veer Narmad South Gujarat University, cyanide present in the soil as sole source of nitrogen and carbon. For the study, cell free extract Surat, Gujarat 395007, India of isolated bacterial species grown in the presence of potassium cyanide (KCN) was prepared and activities of enzymes having role in cyanide degradation pathway were observed. Literature Piyush Desai studies had revealed that mainly four enzymes Cyanide dihydratase, Cyanide oxygenases (mono Department of Biosciences, Veer or dioxygenase), Nitrilase and Cyanase play an important role in different cyanide degrading Narmad South Gujarat University, Surat, Gujarat 395007, India pathways so activity of these four enzymes was detected on the basis of the measurement of released end products. -

Depth-Dependent Transcriptomic Response of Diatoms During Spring
www.nature.com/scientificreports OPEN Depth-dependent transcriptomic response of diatoms during spring bloom in the western subarctic Received: 10 April 2019 Accepted: 24 September 2019 Pacifc Ocean Published: xx xx xxxx Shigekatsu Suzuki1, Takafumi Kataoka2, Tsuyoshi Watanabe3, Haruyo Yamaguchi 1, Akira Kuwata3 & Masanobu Kawachi1 Diatoms play important roles in primary production and carbon transportation in various environments. Large-scale diatom bloom occurs worldwide; however, metabolic responses of diatoms to environmental conditions have been little studied. Here, we targeted the Oyashio region of the western subarctic Pacifc where diatoms bloom every spring and investigated metabolic response of major diatoms to bloom formation by comparing metatranscriptomes between two depths corresponding to diferent bloom phases. Thalassiosira nordenskioeldii and Chaetoceros debilis are two commonly occurring species at the study site. The gene expression profle was drastically diferent between the surface (late decline phase of the bloom; 10 m depth) and the subsurface chlorophyll maximum (SCM, initial decline phase of the bloom; 30 m depth); in particular, both species had high expression of genes for nitrate uptake at the surface, but for ammonia uptake at the SCM. Our culture experiments using T. nordenskioeldii imitating the environmental conditions showed that gene expression for nitrate and ammonia transporters was induced by nitrate addition and active cell division, respectively. These results indicate that the requirement for diferent nitrogen compounds is a major determinant of diatom species responses during bloom maturing. Diatoms contribute up to 20–40% of oceanic primary productivity forming large-scale blooms mainly in coastal and upwelling regions1–3. Tey are key organisms for carbon fux in the ocean, acting as large carbon sinks by ver- tical carbon transportation by sinking to deeper layers4. -

POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii
POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii http://rcin.org.pl WSKAZÓWKI DLA AUTORÓW Kwartalnik „Postępy Biochemii” publikuje artykuły monograficzne omawiające wąskie tematy, oraz artykuły przeglądowe referujące szersze zagadnienia z biochemii i nauk pokrewnych. Artykuły pierwszego typu winny w sposób syntetyczny omawiać wybrany temat na podstawie możliwie pełnego piśmiennictwa z kilku ostatnich lat, a artykuły drugiego typu na podstawie piśmiennictwa z ostatnich dwu lat. Objętość takich artykułów nie powinna przekraczać 25 stron maszynopisu (nie licząc ilustracji i piśmiennictwa). Kwartalnik publikuje także artykuły typu minireviews, do 10 stron maszynopisu, z dziedziny zainteresowań autora, opracowane na podstawie najnow szego piśmiennictwa, wystarczającego dla zilustrowania problemu. Ponadto kwartalnik publikuje krótkie noty, do 5 stron maszynopisu, informujące o nowych, interesujących osiągnięciach biochemii i nauk pokrewnych, oraz noty przybliżające historię badań w zakresie różnych dziedzin biochemii. Przekazanie artykułu do Redakcji jest równoznaczne z oświadczeniem, że nadesłana praca nie była i nie będzie publikowana w innym czasopiśmie, jeżeli zostanie ogłoszona w „Postępach Biochemii”. Autorzy artykułu odpowiadają za prawidłowość i ścisłość podanych informacji. Autorów obowiązuje korekta autorska. Koszty zmian tekstu w korekcie (poza poprawieniem błędów drukarskich) ponoszą autorzy. Artykuły honoruje się według obowiązujących stawek. Autorzy otrzymują bezpłatnie 25 odbitek swego artykułu; zamówienia na dodatkowe odbitki (płatne) należy zgłosić pisemnie odsyłając pracę po korekcie autorskiej. Redakcja prosi autorów o przestrzeganie następujących wskazówek: Forma maszynopisu: maszynopis pracy i wszelkie załączniki należy nadsyłać w dwu egzem plarzach. Maszynopis powinien być napisany jednostronnie, z podwójną interlinią, z marginesem ok. 4 cm po lewej i ok. 1 cm po prawej stronie; nie może zawierać więcej niż 60 znaków w jednym wierszu nie więcej niż 30 wierszy na stronie zgodnie z Normą Polską. -

Niche Strategy of Harmful Algae Revealed Through
SUPPORTING I NFORMATION Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics; Gobler, C.J. et al MATERIALS AND METHODS 1. A. anophagefferens DNA isolation Eight 2-liter flasks with one liter of Aureococcus anophagefferens CCMP1984 culture each were grown axenically to mid-exponential growth phase (107 cells ml-1). The eight flasks were combined in a carboy and harvested with the Sharples continuous flow centrifuge. Cells were scraped from the mylar film, rinsed, and concentrated with a tabletop centrifuge to yield a wet weight of ~ 1 gram. The tube was temporarily stored in liquid N2 vapors. After temporary storage, the bottom half of the tube was placed in a 68º C water bath and heated enough to free the pellet from the tube at which point it was placed in a mortar filled with liquid nitrogen. The pellet was ground using a pestle, and placed into 5 different 50 mL plastic centrifuge tubes, to which Cetyltrimethyl Ammonium Bromide (CTAB) at 68º C was added. Beta-mercaptoethanol (1% final volume) was added to each tube. Tubes were periodically mixed by partial inversion and were incubated for three hours at 68ºC. An equal volume of phenol:chloroform:isoamyl alcohol (25:24:1 ratio) was then added to the cell suspension/CTAB. The tubes were mixed gently by partial inversion and centrifuged in a tabletop centrifuge. The aqueous fraction was removed and an equal volume of chloroform:isoamyl alcohol (24:1) was added and mixed by partial inversion, and then centrifuged. After the second chloroform extraction, the aqueous phase was transferred to a new tube and 0.6 volume of cold (-20º C) isopropanol was added. -

UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Bioinformatic characterization of the 4-Toluene Sulfonate Uptake Permease (TSUP) family of transmembrane proteins : identification of the microbial rhodopsin superfamily and evidence for an ancestral transmembrane hairpin structure Permalink https://escholarship.org/uc/item/5n985064 Author Shlykov, Maksim Aleksandrovich Publication Date 2011 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Bioinformatic Characterization of the 4-Toluene Sulfonate Uptake Permease (TSUP) Family of Transmembrane Proteins: Identification of the Microbial Rhodopsin Superfamily and Evidence for an Ancestral Transmembrane Hairpin Structure A thesis submitted in partial satisfaction of the requirements for the degree Master of Science in Biology by Maksim Aleksandrovich Shlykov Committee in charge: Professor Milton H. Saier, Jr., Chair Professor James W. Golden Professor Emeritus Immo E. Scheffler 2011 ! The Thesis of Maksim Aleksandrovich Shlykov is approved and it is acceptable in quality and form for publication on microfilm and electronically: _________________________________________________________________ _________________________________________________________________ _________________________________________________________________ Chair University of California, San Diego 2011 iii ! DEDICATION I dedicate this thesis to my family, friends, Dr. Saier and all of the previous and current members of Saier Lab who I have had the pleasure of meeting and working with and those whose work made this project possible. This thesis is also dedicated to all of the professors across multiple disciplines, especially biology, who prepared me for this undertaking. A huge thank you is owed to Dorjee Tamang, Abe Silverio, Wei Hao Zheng, Jonathan Chen, Ankur Malhotra, Erik Clarke, Luis Felipe Patino-Cuadrado, Vamsee Reddy and Ming-Ren Yen for their support and guidance. -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000317615Cyc: Dactylococcopsis salina PCC 8305 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari a sulfonate glycine betaine glycine betaine glycine betaine pro pro ammonium ammonium hydrogencarbonate nitrate hydrogencarbonate phosphate phosphate phosphate phosphate phosphate pro pro pro nitrate RS15670 RS08720 RS10615 RS07690 RS05530 RS17475 RS03390 RS06795 RS08115 RS14450 RS15135 RS11065 RS15325 RS07155 RS12795 glycine betaine glycine betaine glycine betaine a sulfonate ammonium ammonium nitrate hydrogencarbonate pro pro pro pro pro hydrogencarbonate phosphate phosphate phosphate phosphate phosphate nitrate 5-amino-5-(4- a reduced an sn-glycero-3- UDP-N-acetyl- an N-terminal- Inorganic Nutrient Metabolism an oxidized a lipid II H + RS00790 H + Amine and Polyamine Fermentation mixed acid fermentation hydroxybenzyl)- SAM + ferredoxin [iron- phospho-N-(acyl) α-D-muramoyl- L-methionyl-L- 3-sulfino-L-alanine thioredoxin H ADP ATP Biosynthesis 6-(D-ribitylimino) sulfur] -
(12) Patent Application Publication (10) Pub. No.: US 2017/0101638 A1 Liao Et Al
US 201701 01638A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0101638 A1 Liao et al. (43) Pub. Date: Apr. 13, 2017 (54) ACETYL-COA CARBOXYLASES (60) Provisional application No. 61/852.387, filed on Mar. 15, 2013. (71) Applicant: Cargill, Incorporated, Wayzata, MN (US) (72) Inventors: Hans Liao, Superior, CO (US); Publication Classification Christopher Patrick Mercogliano, Minneapolis, MN (US); Travis Robert (51) Int. Cl. Wolter, Denver, CO (US); Michael Tai CI2N 9/00 (2006.01) Man Louie, Broomfield, CO (US); (52) U.S. Cl. Wendy Kathleen Ribble, Arvada, CO CPC ........ CI2N 9/93 (2013.01); C12Y 604/01002 (US); Tanya E. W. Lipscomb, Boulder, (2013.01) CO (US); Eileen Colie Spindler, Lafayette, CO (US); Michael D. Lynch, Durham, NC (US) (57) ABSTRACT (21) Appl. No.: 15/269,382 (22) Filed: Sep. 19, 2016 The present invention provides various combinations of genetic modifications to a transformed host cell that provide Related U.S. Application Data increase conversion of carbon to a chemical product. The (63) Continuation of application No. 14/215,379, filed on present invention also provides methods of fermentation and Mar. 17, 2014, now Pat. No. 9,447,438. methods of making various chemical products. Patent Application Publication US 2017/0101638A1 ['OIH Patent Application Publication US 2017/0101638A1 dH-8.OLEICTÄHEGIT\/II/NESHIV/NOTVINHONOISHEIANOO Z*OIH Patent Application Publication US 2017/0101638A1 vo)-?K1938 US 2017/0101638A1 (Oluod 96) A-A ow Patent Application Publication Apr. 13, 2017. Sheet 5 of 9 US 2017/0101638A1 Patent Application Publication Apr. 13, 2017. Sheet 6 of 9 US 2017/0101638A1 o 2 t O S. -
Supplementary Material the Effect of Hyper-Osmotic Salinity on Protein Pattern and Enzyme Activities of Halophytes
10.1071/FP12387_AC © CSIRO 2013 Supplementary Material: Functional Plant Biology, 2013, 40(8–9), 787–804. Supplementary Material The effect of hyper-osmotic salinity on protein pattern and enzyme activities of halophytes Hans-Werner KoyroA, Christian ZörbB, Ahmed DebezC,D and Bernhard HuchzermeyerC,E AInstitute of Plant Ecology, Justus-Liebig-University Giessen, Heinrich-Buff-Ring 26-32, D-35392 Giessen, Germany. BInstitute of Biology, Botany, University Leipzig, Johannisallee 21–23, D-04103 Leipzig, Germany. CInstitut of Botany, Leibniz Universitaet Hannover, Herrenhaeuser-Str. 2, D-30419 Hannover, Germany. DLaboratoire des Plantes Extrêmophiles (LPE), Centre de Biotechnologie à la Technopole de Borj-Cedria (CBBC), BP 901, Hammam-Lif 2050, Tunisia. ECorresponding author. Email: [email protected] Table S1. Identified protein spots No. Name No. Name No. Name 1 Os01g0233000 (DREPP) 71 6-P-gluconate dehydrogenase 141 Myosin heavy chain 2 Anion channel, voltage dependent 72 ATPase family protein 142 DNA Topo-isomerase II 3 K+ channel, voltage gated 73 PPase 143 Annexin (Ca-binding) 4 V-ATPase 74 Os12g0230100 144 Golgi associated proteinse-wap41 5 V-ATPase subunit A 75 Sucrose synthase 145 Glycerophosphodiesterase 6 V-ATPase subunit B 76 Pyruvate dehydrogenase E1-ß 146 Peptidyl-prolyl cis-trans isomerase 7 Chaperonin 77 Pyruvate kinase 147 Protein disulfide-isomerase 8 HSP-20 78 Glucose-6-P isomerase 148 Proteasome domain 9 60 kDa chaperonin alpha 79 ß-glucosidase I 149 Adenosine kinase 10 60 kDa chaperonin alpha 80 Lipoamide -
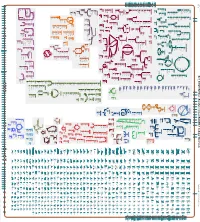
Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000293525Cyc: Bacillus cereus BAG6O-1 Cellular Overview Connections between pathways are omitted for legibility.