Iron-Sulfur Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Impaired Lysosomal Acidification Triggers Iron Deficiency And
RESEARCH ARTICLE Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo King Faisal Yambire1, Christine Rostosky2, Takashi Watanabe3, David Pacheu-Grau1, Sylvia Torres-Odio4, Angela Sanchez-Guerrero1,2, Ola Senderovich5, Esther G Meyron-Holtz5, Ira Milosevic2, Jens Frahm3, A Phillip West4, Nuno Raimundo1* 1Institute of Cellular Biochemistry, University Medical Center Goettingen, Goettingen, Germany; 2European Neuroscience Institute, a Joint Initiative of the Max-Planck Institute and of the University Medical Center Goettingen, Goettingen, Germany; 3Biomedizinische NMR, Max-Planck Institute for Biophysical Chemistry, Goettingen, Germany; 4Department of Microbial Pathogenesis and Immunology, Texas A&M University Health Science Center, Austin, United States; 5Faculty of Biotechnology and Food Engineering, Technion Israel Institute of Technology, Haifa, Israel Abstract Lysosomal acidification is a key feature of healthy cells. Inability to maintain lysosomal acidic pH is associated with aging and neurodegenerative diseases. However, the mechanisms elicited by impaired lysosomal acidification remain poorly understood. We show here that inhibition of lysosomal acidification triggers cellular iron deficiency, which results in impaired mitochondrial function and non-apoptotic cell death. These effects are recovered by supplying iron via a lysosome-independent pathway. Notably, iron deficiency is sufficient to trigger inflammatory signaling in cultured primary neurons. Using a mouse model of impaired lysosomal acidification, we observed a robust iron deficiency response in the brain, verified by in vivo magnetic resonance *For correspondence: imaging. Furthermore, the brains of these mice present a pervasive inflammatory signature [email protected] associated with instability of mitochondrial DNA (mtDNA), both corrected by supplementation of goettingen.de the mice diet with iron. Our results highlight a novel mechanism linking impaired lysosomal Competing interests: The acidification, mitochondrial malfunction and inflammation in vivo. -

Effect of Genotype on Micronutrient Absorption and Metabolism: a Review of Iron, Copper, Iodine and Selenium, and Folates Richard Mithen
Int. J. Vitam. Nutr. Res., 77 (3), 2007, 205–216 Effect of Genotype on Micronutrient Absorption and Metabolism: a Review of Iron, Copper, Iodine and Selenium, and Folates Richard Mithen Institute of Food Research, Colney Lane, Norwich, NR4 7UA, UK Received for publication: July 28, 2006 Abstract: For the majority of micronutrients, there are very little data, or none at all, on the role of genetic poly- morphisms on their absorption and metabolism. In many cases, the elucidation of biochemical pathways and regulators of homeostatic mechanisms have come from studies of individuals that have mutations in certain genes. Other polymorphisms in these genes that result in a less severe phenotype may be important in determining the natural range of variation in absorption and metabolism that is commonly observed. To illustrate some of these aspects, I briefly review the increased understanding of iron metabolism that has arisen from our knowledge of the effects of mutations in several genes, the role of genetic variation in mediating the nutritional effects of io- dine and selenium, and finally, the interaction between a genetic polymorphism in folate metabolism and folic acid fortification. Key words: Micronutrients, genetic polymorphisms, iron, iodine, selenium, folates Introduction the interpretation of epidemiological studies, in which some of the variation observed in nutrient status or re- Recently there has been considerable interest in the role quirement may be due to genetic variation at a few or sev- that genetic polymorphisms may play in several aspects eral loci that determine the uptake and metabolism of var- of human nutrition, and the ill-defined terms nutrige- ious nutrients. -

Small-Molecule Binding Sites to Explore New Targets in the Cancer Proteome
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2016 Small-molecule binding sites to explore new targets in the cancer proteome David Xu, Shadia I. Jalal, George W. Sledge Jr., and Samy O. Meroueh* Supplementary Text Druggable Binding Sites across all 10 Diseases. Using the previously established cutoffs, we identified genes that were overexpressed across multiple cancer types and featured druggable binding sites. We ranked these genes based on the total number of tumors that overexpressed the gene (Fig. S1). Using a simple PubMed query, we then counted the number of articles in which either the gene symbol or gene name was co-mentioned with the term ‘cancer’. Most of the most frequently occurring differentially-expressed genes correspond to proteins of well- established cancer targets. Among them are matrix metalloproteinases (MMPs), including MMP1, MMP9, and MMP12, which are implicated in tumor invasion and metastasis (1). There are several protein kinases, including TTK, AURKA, AURKB, and PLK1, that are involved in cell signaling and well-established oncology targets (2). Some genes among this list that have not been extensively studied nor targeted in cancer. These include the serine/threonine kinase PKMYT1 (MYT1) is a regulator of G2/M transition in the cell cycle, but lacks focused small molecule inhibitors that specifically target the kinase. Recent efforts in developing small molecule inhibitors involve repurposing of available kinase inhibitors to specifically target the kinase (3). A subunit of the GINS complex GINS2 (PSF2) is involved in cell proliferation and survival in cancer cell lines (4,5). -

Oxidized Phospholipids Regulate Amino Acid Metabolism Through MTHFD2 to Facilitate Nucleotide Release in Endothelial Cells
ARTICLE DOI: 10.1038/s41467-018-04602-0 OPEN Oxidized phospholipids regulate amino acid metabolism through MTHFD2 to facilitate nucleotide release in endothelial cells Juliane Hitzel1,2, Eunjee Lee3,4, Yi Zhang 3,5,Sofia Iris Bibli2,6, Xiaogang Li7, Sven Zukunft 2,6, Beatrice Pflüger1,2, Jiong Hu2,6, Christoph Schürmann1,2, Andrea Estefania Vasconez1,2, James A. Oo1,2, Adelheid Kratzer8,9, Sandeep Kumar 10, Flávia Rezende1,2, Ivana Josipovic1,2, Dominique Thomas11, Hector Giral8,9, Yannick Schreiber12, Gerd Geisslinger11,12, Christian Fork1,2, Xia Yang13, Fragiska Sigala14, Casey E. Romanoski15, Jens Kroll7, Hanjoong Jo 10, Ulf Landmesser8,9,16, Aldons J. Lusis17, 1234567890():,; Dmitry Namgaladze18, Ingrid Fleming2,6, Matthias S. Leisegang1,2, Jun Zhu 3,4 & Ralf P. Brandes1,2 Oxidized phospholipids (oxPAPC) induce endothelial dysfunction and atherosclerosis. Here we show that oxPAPC induce a gene network regulating serine-glycine metabolism with the mitochondrial methylenetetrahydrofolate dehydrogenase/cyclohydrolase (MTHFD2) as a cau- sal regulator using integrative network modeling and Bayesian network analysis in human aortic endothelial cells. The cluster is activated in human plaque material and by atherogenic lipo- proteins isolated from plasma of patients with coronary artery disease (CAD). Single nucleotide polymorphisms (SNPs) within the MTHFD2-controlled cluster associate with CAD. The MTHFD2-controlled cluster redirects metabolism to glycine synthesis to replenish purine nucleotides. Since endothelial cells secrete purines in response to oxPAPC, the MTHFD2- controlled response maintains endothelial ATP. Accordingly, MTHFD2-dependent glycine synthesis is a prerequisite for angiogenesis. Thus, we propose that endothelial cells undergo MTHFD2-mediated reprogramming toward serine-glycine and mitochondrial one-carbon metabolism to compensate for the loss of ATP in response to oxPAPC during atherosclerosis. -

An Insight Into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses Chontida Yarana University of Kentucky, [email protected]
University of Kentucky UKnowledge Toxicology and Cancer Biology Faculty Toxicology and Cancer Biology Publications 9-28-2017 Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses Chontida Yarana University of Kentucky, [email protected] Daret K. St. Clair University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits oy u. Follow this and additional works at: https://uknowledge.uky.edu/toxicology_facpub Part of the Medical Toxicology Commons Repository Citation Yarana, Chontida and St. Clair, Daret K., "Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles- Mediated Oxidative Stress Responses" (2017). Toxicology and Cancer Biology Faculty Publications. 62. https://uknowledge.uky.edu/toxicology_facpub/62 This Review is brought to you for free and open access by the Toxicology and Cancer Biology at UKnowledge. It has been accepted for inclusion in Toxicology and Cancer Biology Faculty Publications by an authorized administrator of UKnowledge. For more information, please contact [email protected]. Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses Notes/Citation Information Published in Antioxidants, v. 6, issue 4, 75, p. 1-17. © 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/). Digital Object Identifier (DOI) https://doi.org/10.3390/antiox6040075 This review is available at UKnowledge: https://uknowledge.uky.edu/toxicology_facpub/62 antioxidants Review Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses Chontida Yarana 1,2 and Daret K. -

STEAP Proteins: from Structure to Applications in Cancer Therapy
Published OnlineFirst April 20, 2012; DOI: 10.1158/1541-7786.MCR-11-0281 Molecular Cancer Review Research STEAP Proteins: From Structure to Applications in Cancer Therapy Ines^ M. Gomes, Claudio J. Maia, and Cecília R. Santos Abstract The human 6-transmembrane epithelial antigen of prostate (STEAP) family comprises STEAP1, STEAP2, STEAP3, and STEAP4. All of these proteins are unique to mammals and share an innate activity as metallor- eductases, indicating their importance in metal metabolism. Overall, they participate in a wide range of biologic processes, such as molecular trafficking in the endocytic and exocytic pathways and control of cell proliferation and apoptosis. STEAP1 and STEAP2 are overexpressed in several types of human cancers, namely prostate, bladder, colon, pancreas, ovary, testis, breast, cervix, and Ewing sarcoma, but their clinical significance and role in cancer cells are not clear. Still, their localization in the cell membrane and differential expression in normal and cancer tissues make STEAP proteins potential candidates as biomarkers of several cancers, as well as potential targets for new immunotherapeutic strategies for disease attenuation or treatment. This review brings together the current knowledge about each STEAP protein, giving an overview of the roles of this family of proteins in human physiology and disease, and analyzes their potential as immunotherapeutic agents in cancer research. Mol Cancer Res; 1–15. Ó2012 AACR. Introduction bic amino acid) that is responsible for targeting transmem- The 6-transmembrane epithelial antigen of prostate braneproteinstolysosomesandendosomes,andtheRossman fold (GXGXXG/A motif), a feature of proteins with oxido- (STEAP) family of proteins includes 4 members, named fi 6-transmembrane epithelial antigen of prostate 1 to 4 reductase and dehydrogenase functions (3, 5). -

SUPPLEMENTARY APPENDIX Exome Sequencing Reveals Heterogeneous Clonal Dynamics in Donor Cell Myeloid Neoplasms After Stem Cell Transplantation
SUPPLEMENTARY APPENDIX Exome sequencing reveals heterogeneous clonal dynamics in donor cell myeloid neoplasms after stem cell transplantation Julia Suárez-González, 1,2 Juan Carlos Triviño, 3 Guiomar Bautista, 4 José Antonio García-Marco, 4 Ángela Figuera, 5 Antonio Balas, 6 José Luis Vicario, 6 Francisco José Ortuño, 7 Raúl Teruel, 7 José María Álamo, 8 Diego Carbonell, 2,9 Cristina Andrés-Zayas, 1,2 Nieves Dorado, 2,9 Gabriela Rodríguez-Macías, 9 Mi Kwon, 2,9 José Luis Díez-Martín, 2,9,10 Carolina Martínez-Laperche 2,9* and Ismael Buño 1,2,9,11* on behalf of the Spanish Group for Hematopoietic Transplantation (GETH) 1Genomics Unit, Gregorio Marañón General University Hospital, Gregorio Marañón Health Research Institute (IiSGM), Madrid; 2Gregorio Marañón Health Research Institute (IiSGM), Madrid; 3Sistemas Genómicos, Valencia; 4Department of Hematology, Puerta de Hierro General University Hospital, Madrid; 5Department of Hematology, La Princesa University Hospital, Madrid; 6Department of Histocompatibility, Madrid Blood Centre, Madrid; 7Department of Hematology and Medical Oncology Unit, IMIB-Arrixaca, Morales Meseguer General University Hospital, Murcia; 8Centro Inmunológico de Alicante - CIALAB, Alicante; 9Department of Hematology, Gregorio Marañón General University Hospital, Madrid; 10 Department of Medicine, School of Medicine, Com - plutense University of Madrid, Madrid and 11 Department of Cell Biology, School of Medicine, Complutense University of Madrid, Madrid, Spain *CM-L and IB contributed equally as co-senior authors. Correspondence: -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Cellular Iron Homeostasis
BioIron 2017 Educational Session Cellular iron metabolism Kostas Pantopoulos Lady Davis Institute for Medical Research Division of Experimental Medicine Montreal, Quebec, Canada The problem of iron acquisition Iron is an abundant metal, but… In aqueous solutions, Fe2+ is readily oxidized to Fe3+ 3+ At neutral pH, Fe forms essentially insoluble Fe(OH)3 Iron’s bioavailability is limited Strategies for iron acquisition (1): direct iron transport 1. Reduction of Fe3+ to Fe2+ in the vicinity of the cell by plasma membrane-bound ferric reductases Frep Fre reductases 2. Direct uptake by low affinity (Km=40 mM) ferrous transporter Fet4p or Reoxidation by plasma membrane-bound ferroxidase Fet3p and internalization by permease Ftr1p in a high affinity transporting system (K =0.15 mM) Fet3 oxidase/Ftr1 permease m Strategies for iron acquisition (2): indirect iron uptake Many bacteria and lower eukaryotes secrete siderophores (low molecular weight Fe3+ chelators) Iron-loaded siderophores are internalized into the bacteria by binding to specific cell-surface receptors Deferroxamine, a bacterial siderophore, is widely employed for iron chelation therapy deferroxamine (desferral) Iron assimilation in higher organisms Both concepts: Direct iron transport by trans-membrane transporters, following reduction to soluble Fe2+ and Receptor-mediated uptake of iron, that is “captured” in form of Fe3+ by an iron-binding molecule have been conserved in higher organisms Direct iron transport is critical for dietary iron absorption Iron absorption from intestinal -
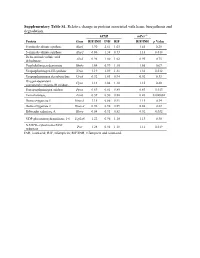
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator- -

Structure of the Membrane Proximal Oxidoreductase Domain of Human Steap3, the Dominant Ferrireductase of the Erythroid Transferrin Cycle
Structure of the membrane proximal oxidoreductase domain of human Steap3, the dominant ferrireductase of the erythroid transferrin cycle Anoop K. Sendamarai*, Robert S. Ohgami†, Mark D. Fleming†, and C. Martin Lawrence*‡ *Department of Chemistry and Biochemistry, Montana State University, Bozeman, MT 59717; and †Department of Pathology, Children’s Hospital and Harvard Medical School, 300 Longwood Avenue, Boston, MA 02115 Edited by Pamela J. Bjorkman, California Institute of Technology, Pasadena, CA, and approved March 20, 2008 (received for review February 8, 2008) The daily production of 200 billion erythrocytes requires 20 mg of undergo phagocytosis by macrophages, and much of the eryth- iron, accounting for nearly 80% of the iron demand in humans. rocyte iron is recycled, drastically reducing the need for dietary Thus, erythroid precursor cells possess an efficient mechanism for uptake of additional iron (9). To meet their iron need, erythroid iron uptake in which iron loaded transferrin (Tf) binds to the precursor cells are uniquely dependent on the transferrin cycle transferrin receptor (TfR) at the cell surface. The Tf:TfR complex (4, 10). In this cycle, ferric (Fe3ϩ) iron-loaded transferrin (Tf) then enters the endosome via receptor-mediated endocytosis. binds to the transferrin receptor (TfR-1) on the cell surface. The Upon endosomal acidification, iron is released from Tf, reduced to Tf:TfR-1 complex then enters the endosome via receptor- Fe2؉ by Steap3, and transported across the endosomal membrane mediated endocytosis. Within the endosome, iron is released by divalent metal iron transporter 1. Steap3, the major ferrireduc- from Tf and then is reduced from Fe3ϩ to Fe2ϩ by Steap3, tase in erythrocyte endosomes, is a member of a unique family of permitting transport across the endosomal membrane by diva- reductases. -

Studies of the Structure and Function of Recombinant Human Hephaestin
STUDIES OF THE STRUCTURE AND FUNCTION OF RECOMBINANT HUMAN HEPHAESTIN by Ganna Vashchenko B.Sc., Taras Shevchenko National University of Kyiv, 2007 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Biochemistry and Molecular Biology) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) July 2012 © Ganna Vashchenko, 2012 ABSTRACT Hephaestin is a multicopper ferroxidase involved in iron absorption in the small intestine. The ferroxidase activity of hephaestin is thought to play an important role during iron export from intestinal enterocytes and the subsequent iron loading of the blood protein transferrin, which delivers iron to the tissues. Structurally, the ectodomain of hephaestin is predicted to resemble ceruloplasmin, the soluble ferroxidase of blood. In this work I investigated substrate specificity, copper loading and the ferroxidation mechanism of recombinantly expressed human hephaestin. The hephaestin ectodomain (Fet3Hp) was expressed in Pichia pastoris and purified to electrophoretic homogeneity by immunoaffinity chromatography. Recombinant hephaestin retained ferroxidase activity and showed an average copper content of 4.2 copper atoms per molecule. The Km values of Fet3Hp for such organic substrates as p-phenylenediamine and o- dianisidine were close to values determined for ceruloplasmin. However, in contrast to ceruloplasmin, recombinant hephaestin was incapable of direct oxidation of adrenaline and dopamine implying a difference in biological substrate specificities between these two homologous oxidases. I also expressed hephaestin ectodomain with the ceruloplasmin signal peptide (CpHp) using BHK cells as an expression system. Ion exchange chromatography of purified CpHp resulted in the production of a hephaestin fraction with improved catalytic and spectroscopic properties.