University of California, San Diego
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Role of 15-Lipoxygenase/15-Hydroxyeicosatetraenoic Acid in Hypoxia-Induced Pulmonary Hypertension
J Physiol Sci (2012) 62:163–172 DOI 10.1007/s12576-012-0196-9 REVIEW Role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in hypoxia-induced pulmonary hypertension Daling Zhu • Yajuan Ran Received: 29 September 2011 / Accepted: 25 January 2012 / Published online: 14 February 2012 Ó The Physiological Society of Japan and Springer 2012 Abstract Pulmonary arterial hypertension (PAH) is a Introduction rare disease with a complex aetiology characterized by elevated pulmonary artery resistance, which leads to right Pulmonary hypertension (PH) is a severe and frequently heart ventricular afterload and ultimately progressing to fatal disease characterized by elevated mean pulmonary right ventricular failure and often death. In addition to arterial (PA) pressure greater than 25 mmHg at rest or other factors, metabolites of arachidonic acid cascade play greater than 30 mmHg with exercise [1], and which con- an important role in the pulmonary vasculature, and dis- tributes to the morbidity and mortality of adult and pedi- ruption of signaling pathways of arachidonic acid plays a atric patients with various lung and heart diseases. central role in the pathogenesis of PAH. 15-Lipoxygenase According to the Venice Classification of Pulmonary (15-LO) is upregulated in pulmonary artery endothelial Hypertension in 2003, PH is currently classified into five cells and smooth muscle cells of PAH patients, and its categories as listed in Table 1. Importantly, many of these metabolite 15-hydroxyeicosatetraenoic acid (15-HETE) in diseases or conditions are associated with persistent or particular seems to play a central role in the contractile intermittent hypoxia, either globally or regionally, within machinery, and in the initiation and propagation of cell confined areas of the lung [2]. -

Effects of Glycosylation on the Enzymatic Activity and Mechanisms of Proteases
International Journal of Molecular Sciences Review Effects of Glycosylation on the Enzymatic Activity and Mechanisms of Proteases Peter Goettig Structural Biology Group, Faculty of Molecular Biology, University of Salzburg, Billrothstrasse 11, 5020 Salzburg, Austria; [email protected]; Tel.: +43-662-8044-7283; Fax: +43-662-8044-7209 Academic Editor: Cheorl-Ho Kim Received: 30 July 2016; Accepted: 10 November 2016; Published: 25 November 2016 Abstract: Posttranslational modifications are an important feature of most proteases in higher organisms, such as the conversion of inactive zymogens into active proteases. To date, little information is available on the role of glycosylation and functional implications for secreted proteases. Besides a stabilizing effect and protection against proteolysis, several proteases show a significant influence of glycosylation on the catalytic activity. Glycans can alter the substrate recognition, the specificity and binding affinity, as well as the turnover rates. However, there is currently no known general pattern, since glycosylation can have both stimulating and inhibiting effects on activity. Thus, a comparative analysis of individual cases with sufficient enzyme kinetic and structural data is a first approach to describe mechanistic principles that govern the effects of glycosylation on the function of proteases. The understanding of glycan functions becomes highly significant in proteomic and glycomic studies, which demonstrated that cancer-associated proteases, such as kallikrein-related peptidase 3, exhibit strongly altered glycosylation patterns in pathological cases. Such findings can contribute to a variety of future biomedical applications. Keywords: secreted protease; sequon; N-glycosylation; O-glycosylation; core glycan; enzyme kinetics; substrate recognition; flexible loops; Michaelis constant; turnover number 1. -

The Relationship Between Specialized Pro-Resolving Lipid Mediators, Morbid Obesity and Weight Loss After Bariatric Surgery
www.nature.com/scientificreports OPEN The relationship between specialized pro‑resolving lipid mediators, morbid obesity and weight loss after bariatric surgery Fabian Schulte1,2,7, Abdul Aziz Asbeutah3,6,7, Peter N. Benotti4, G. Craig Wood4, Christopher Still4, Bruce R. Bistrian5, Markus Hardt1,2 & Francine K. Welty3* Obesity and diabetes are associated with chronic infammation. Specialized pro‑resolving lipid mediators (SPMs)—resolvins (Rv), protectins (PD) and maresins (MaR)—actively resolve infammation. Bariatric surgery achieves remission of diabetes, but mechanisms are unclear. We measured SPMs and proinfammatory eicosanoid levels using liquid chromatography‑tandem mass spectrometry in 29 morbidly obese subjects (13 with diabetes) and 15 nondiabetic, mildly obese subjects. Compared to the mildly obese, the morbidly obese had higher levels of SPMs—RvD3, RvD4 and PD1—and white blood cells (WBC) and platelets. Post‑surgery, SPM and platelet levels decreased in morbidly obese nondiabetic subjects but not in diabetic subjects, suggesting continued infammation. Despite similar weight reductions 1 year after surgery (44.6% vs. 46.6%), 8 diabetes remitters had signifcant reductions in WBC and platelet counts whereas fve non‑remitters did not. Remitters had a 58.2% decrease (p = 0.03) in 14‑HDHA, a maresin pathway marker; non‑remitters had an 875.7% increase in 14‑HDHA but a 36.9% decrease in MaR1 to a median of 0. In conclusion, higher levels of RvD3, PD1 and their pathway marker, 17‑HDHA, are markers of leukocyte activation and infammation in morbid obesity and diabetes and diminish with weight loss in nondiabetic but not diabetic subjects, possibly representing sustained infammation in the latter. -

MALE Protein Name Accession Number Molecular Weight CP1 CP2 H1 H2 PDAC1 PDAC2 CP Mean H Mean PDAC Mean T-Test PDAC Vs. H T-Test
MALE t-test t-test Accession Molecular H PDAC PDAC vs. PDAC vs. Protein Name Number Weight CP1 CP2 H1 H2 PDAC1 PDAC2 CP Mean Mean Mean H CP PDAC/H PDAC/CP - 22 kDa protein IPI00219910 22 kDa 7 5 4 8 1 0 6 6 1 0.1126 0.0456 0.1 0.1 - Cold agglutinin FS-1 L-chain (Fragment) IPI00827773 12 kDa 32 39 34 26 53 57 36 30 55 0.0309 0.0388 1.8 1.5 - HRV Fab 027-VL (Fragment) IPI00827643 12 kDa 4 6 0 0 0 0 5 0 0 - 0.0574 - 0.0 - REV25-2 (Fragment) IPI00816794 15 kDa 8 12 5 7 8 9 10 6 8 0.2225 0.3844 1.3 0.8 A1BG Alpha-1B-glycoprotein precursor IPI00022895 54 kDa 115 109 106 112 111 100 112 109 105 0.6497 0.4138 1.0 0.9 A2M Alpha-2-macroglobulin precursor IPI00478003 163 kDa 62 63 86 72 14 18 63 79 16 0.0120 0.0019 0.2 0.3 ABCB1 Multidrug resistance protein 1 IPI00027481 141 kDa 41 46 23 26 52 64 43 25 58 0.0355 0.1660 2.4 1.3 ABHD14B Isoform 1 of Abhydrolase domain-containing proteinIPI00063827 14B 22 kDa 19 15 19 17 15 9 17 18 12 0.2502 0.3306 0.7 0.7 ABP1 Isoform 1 of Amiloride-sensitive amine oxidase [copper-containing]IPI00020982 precursor85 kDa 1 5 8 8 0 0 3 8 0 0.0001 0.2445 0.0 0.0 ACAN aggrecan isoform 2 precursor IPI00027377 250 kDa 38 30 17 28 34 24 34 22 29 0.4877 0.5109 1.3 0.8 ACE Isoform Somatic-1 of Angiotensin-converting enzyme, somaticIPI00437751 isoform precursor150 kDa 48 34 67 56 28 38 41 61 33 0.0600 0.4301 0.5 0.8 ACE2 Isoform 1 of Angiotensin-converting enzyme 2 precursorIPI00465187 92 kDa 11 16 20 30 4 5 13 25 5 0.0557 0.0847 0.2 0.4 ACO1 Cytoplasmic aconitate hydratase IPI00008485 98 kDa 2 2 0 0 0 0 2 0 0 - 0.0081 - 0.0 -

Phospholipase A2 Regulates Eicosanoid Class Switching During Inflammasome Activation
Phospholipase A2 regulates eicosanoid class switching during inflammasome activation Paul C. Norrisa, David Gosselinb, Donna Reichartb, Christopher K. Glassb, and Edward A. Dennisa,1 Departments of aChemistry/Biochemistry and Pharmacology, and bCellular and Molecular Medicine, University of California, San Diego, La Jolla, CA 92093 Edited by Michael A. Marletta, The Scripps Research Institute, La Jolla, CA, and approved July 30, 2014 (received for review March 13, 2014) Initiation and resolution of inflammation are considered to be to initiate pathogenic killing, subsequent “class switching” to lipoxin tightly connected processes. Lipoxins (LX) are proresolution lipid (LX) formation by “reprogrammed” neutrophils inhibits additional mediators that inhibit phlogistic neutrophil recruitment and pro- neutrophil recruitment during self-resolving inflammatory resolu- mote wound-healing macrophage recruitment in humans via tion (9). The direct link between inflammatory commitment and potent and specific signaling through the LXA4 receptor (ALX). resolution mediated by eicosanoid signaling in macrophages One model of lipoxin biosynthesis involves sequential metabolism remains unclear from short-term vs. long-term priming, but the of arachidonic acid by two cell types expressing a combined trans- complete temporal changes and important interconnections cellular metabolon. It is currently unclear how lipoxins are effi- within the entire eicosadome are now demonstrated. ciently formed from precursors or if they are directly generated after receptor-mediated inflammatory commitment. Here, we pro- Results vide evidence for a pathway by which lipoxins are generated in We first primed immortalized macrophage-like cells (RAW264.7) macrophages as a consequence of sequential activation of toll-like with the TLR4 agonist Kdo2 lipid A (KLA) for various times and receptor 4 (TLR4), a receptor for endotoxin, and P2X7, a purinergic examined the effects on subsequent purinergic stimulated COX receptor for extracellular ATP. -

Nutrition Bytes
UCLA Nutrition Bytes Title Theories Presented in The Zone Permalink https://escholarship.org/uc/item/9pg2j7wk Journal Nutrition Bytes, 3(1) ISSN 1548-4327 Author Chang, Jeannie Publication Date 1997 Peer reviewed eScholarship.org Powered by the California Digital Library University of California "Easy -to -follow" diet plans appear each year to convince overweight Americans that there is finally a way to lose weight permanently. Barry Sears and Bill Lawren’s book, The Zone, is no exception; in fact, Sears’ book cover advertises that it is "a dietary road map to lose weight permanently, reset your genetic code, prevent disease, achieve maximum physical performance, [and] enhance mental productivity." But is this realistic? Can a diet really accomplish all of those tasks? After numerous searches through several databases, only two journal articles analyzing this diet plan surfaced, both warning readers about the misinformation contained in the book. There are definitely problems with Sears’ diet; for example, his daily caloric intake recommendation is dangerously low. Tufts University Diet and Nutrition Letter stated that they believed "[Sears was] confusing the near -euphoria he promises from following the Zone diet with lightheadedness from hunger" (3). Dr. Zamenhof, Associate Professor of Biological Ch emistry at UCLA said that it was probably due to an increased release of endorphins, enkephalins, and catecholamines. In addition, the information he provides regarding carbohydrate and fat metabolism, as well as insulin and glucagon secretion, are wrong. However, Sears also says that there is a direct relationship between insulin and glucagon with the synthesis of "good" eicosanoids (prostaglandin E1 and prostaglandin I2 and "bad" eicosanoids (prostaglandin E2, thromboxanes, and leukotrienes). -

Novel Proteins Regulated by Mtor in Subependymal Giant Cell Astrocytomas of Patients with Tuberous Sclerosis Complex and New Therapeutic Implications
The American Journal of Pathology, Vol. 176, No. 4, April 2010 Copyright © American Society for Investigative Pathology DOI: 10.2353/ajpath.2010.090950 Molecular Pathogenesis of Genetic and Inherited Diseases Novel Proteins Regulated by mTOR in Subependymal Giant Cell Astrocytomas of Patients with Tuberous Sclerosis Complex and New Therapeutic Implications Magdalena Ewa Tyburczy,* Katarzyna Kotulska,† and demonstrated an effective modulation of SEGA Piotr Pokarowski,‡ Jakub Mieczkowski,* growth by pharmacological inhibition of both Joanna Kucharska,* Wieslawa Grajkowska,† mTOR and extracellular signal-regulated kinase sig- Maciej Roszkowski,§ Sergiusz Jozwiak,† naling pathways, which could represent a novel ther- and Bozena Kaminska* apeutic approach. (Am J Pathol 2010, 176:1878–1890; DOI: 10.2353/ajpath.2010.090950) From the Laboratory of Transcription Regulation,* the Nencki Institute of Experimental Biology, Warsaw; the Departments of Neurology and Epileptology,† and Neurosurgery,§ the Children’s Subependymal giant cell astrocytomas (SEGAs) are rare, Memorial Health Institute, Warsaw; and the Faculty of low-grade brain tumors (World Health Organization Grade I) Mathematics, Informatics, and Mechanics,‡ University of of a mixed glioneuronal lineage.1,2 They are observed in Warsaw, Poland 10% to 20% of patients with tuberous sclerosis complex (TSC) and are the major cause of morbidity in children and young adults with TSC.3 The disease affects about one in Subependymal giant cell astrocytomas (SEGAs) are 6000 people, is characterized by the formation of benign rare brain tumors associated with tuberous sclerosis tumors in multiple organs (mainly brain, heart, kidneys, skin, complex (TSC), a disease caused by mutations in TSC1 or lungs), and is often associated with epilepsy, mental or TSC2, resulting in enhancement of mammalian retardation, and autism.4,5 Tuberous sclerosis complex is target of rapamycin (mTOR) activity, dysregulation of caused by mutation in one of two tumor suppressor genes, cell growth, and tumorigenesis. -

Omega-3, Omega-6 and Omega-9 Fatty Acids
Johnson and Bradford, J Glycomics Lipidomics 2014, 4:4 DOI: 0.4172/2153-0637.1000123 Journal of Glycomics & Lipidomics Review Article Open Access Omega-3, Omega-6 and Omega-9 Fatty Acids: Implications for Cardiovascular and Other Diseases Melissa Johnson1* and Chastity Bradford2 1College of Agriculture, Environment and Nutrition Sciences, Tuskegee University, Tuskegee, Alabama, USA 2Department of Biology, Tuskegee University, Tuskegee, Alabama, USA Abstract The relationship between diet and disease has long been established, with epidemiological and clinical evidence affirming the role of certain dietary fatty acid classes in disease pathogenesis. Within the same class, different fatty acids may exhibit beneficial or deleterious effects, with implications on disease progression or prevention. In conjunction with other fatty acids and lipids, the omega-3, -6 and -9 fatty acids make up the lipidome, and with the conversion and storage of excess carbohydrates into fats, transcendence of the glycome into the lipidome occurs. The essential omega-3 fatty acids are typically associated with initiating anti-inflammatory responses, while omega-6 fatty acids are associated with pro-inflammatory responses. Non-essential, omega-9 fatty acids serve as necessary components for other metabolic pathways, which may affect disease risk. These fatty acids which act as independent, yet synergistic lipid moieties that interact with other biomolecules within the cellular ecosystem epitomize the critical role of these fatty acids in homeostasis and overall health. This review focuses on the functional roles and potential mechanisms of omega-3, omega-6 and omega-9 fatty acids in regard to inflammation and disease pathogenesis. A particular emphasis is placed on cardiovascular disease, the leading cause of morbidity and mortality in the United States. -

Inflammation, Cancer and Oxidative Lipoxygenase Activity Are Intimately Linked
Cancers 2014, 6, 1500-1521; doi:10.3390/cancers6031500 OPEN ACCESS cancers ISSN 2072-6694 www.mdpi.com/journal/cancers Review Inflammation, Cancer and Oxidative Lipoxygenase Activity are Intimately Linked Rosalina Wisastra and Frank J. Dekker * Pharmaceutical Gene Modulation, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +31-5-3638030; Fax: +31-5-3637953. Received: 16 April 2014; in revised form: 27 June 2014 / Accepted: 2 July 2014 / Published: 17 July 2014 Abstract: Cancer and inflammation are intimately linked due to specific oxidative processes in the tumor microenvironment. Lipoxygenases are a versatile class of oxidative enzymes involved in arachidonic acid metabolism. An increasing number of arachidonic acid metabolites is being discovered and apart from their classically recognized pro-inflammatory effects, anti-inflammatory effects are also being described in recent years. Interestingly, these lipid mediators are involved in activation of pro-inflammatory signal transduction pathways such as the nuclear factor κB (NF-κB) pathway, which illustrates the intimate link between lipid signaling and transcription factor activation. The identification of the role of arachidonic acid metabolites in several inflammatory diseases led to a significant drug discovery effort around arachidonic acid metabolizing enzymes. However, to date success in this area has been limited. This might be attributed to the lack of selectivity of the developed inhibitors and to a lack of detailed understanding of the functional roles of arachidonic acid metabolites in inflammatory responses and cancer. -

Recombinant Human Dipeptidase 1 Protein
Leader in Biomolecular Solutions for Life Science Recombinant Human Dipeptidase 1 Protein Catalog No.: RP01097 Recombinant Sequence Information Background Species Gene ID Swiss Prot This protein is a kidney membrane enzyme involved in the metabolism of Human 1800 P16444 glutathione and other similar proteins by dipeptide hydrolysis. The encoded protein is known to regulate leukotriene activity by catalyzing the conversion of Tags leukotriene D4 to leukotriene E4. This protein uses zinc as a cofactor and acts as a C-6×His disulfide-linked homodimer. Two transcript variants encoding the same protein have been found for this gene. Synonyms Dipeptidase 1; Dehydropeptidase-I; Microsomal Dipeptidase; Renal Basic Information Dipeptidase; hRDP; DPEP1; MDP; RDP Description Recombinant Human Dipeptidase 1 Protein is produced by Mammalian expression Product Information system. The target protein is expressed with sequence (Asp17-Ser385) of human Dipeptidase 1 (Accession #P16444) fused with a 6×His tag at the C-terminus. Source Purification Mammalian > 95% by SDS- Bio-Activity PAGE. Storage Endotoxin Store the lyophilized protein at -20°C to -80 °C for long term. < 1.0 EU/μg of the protein by LAL After reconstitution, the protein solution is stable at -20 °C for 3 months, at 2-8 °C method. for up to 1 week. Avoid repeated freeze/thaw cycles. Formulation Lyophilized from a 0.22 μm filtered solution of 20mM PB, 150mM NaCl, pH7.4.Contact us for customized product form or formulation. Reconstitution Reconstitute to a concentration of 0.1-0.5 mg/mL in sterile distilled water. Contact www.abclonal.com Validation Data Recombinant Human Dipeptidase 1 Protein was determined by SDS-PAGE with Coomassie Blue, showing a band at 41 kDa. -

The Effects of Omega-3 Fatty Acid Supplementation in Pregnancy on Maternal Eicosanoid, Cytokine, and Chemokine Secretion
0031-3998/09/6602-0212 Vol. 66, No. 2, 2009 PEDIATRIC RESEARCH Printed in U.S.A. Copyright © 2009 International Pediatric Research Foundation, Inc. The Effects of Omega-3 Fatty Acid Supplementation in Pregnancy on Maternal Eicosanoid, Cytokine, and Chemokine Secretion KRISTINA WARSTEDT, CATRIN FURUHJELM, KAREL DUCHE´ N, KARIN FA¨ LTH-MAGNUSSON, AND MALIN FAGERÅS Department of Clinical and Experimental Medicine [K.W., C.F., K.D., K.F.-M., M.F.], Clinical and Experimental Research [K.W., M.F.], Linko¨ping University, Linko¨ping, Sweden ABSTRACT: The incidence of allergic diseases has increased, and i.e. eicosapentaenoic acid (EPA) and docosahexaenoic acid a relation between allergy and dietary fatty acids has been proposed. (DHA), may dampen the proinflammatory effects of AA at Modulation of the maternal immune function during pregnancy may multiple levels (3). -3 LCPUFA supplementation may result have an impact on future clinical outcomes in the child. The aim of in decreased production of AA-derived eicosanoids (3) and this study was to determine the effects of omega ( )-3 long-chain has been reported to decrease lipopolysaccharide (LPS)- polyunsaturated fatty acids (LCPUFA) supplementation during preg- induced interleukin (IL)-1, IL-6, and tumor necrosis factor nancy on the plasma fatty acid composition in relation to the maternal immune function. Pregnant women with allergic disease in their (TNF) production by mononuclear cells (5). immediate family were supplemented daily with 2.7 g -3 LCPUFA Epidemiologic studies suggest that the period from early (n ϭ 70) or 2.8 g soybean oil as placebo (n ϭ 75) from the 25th fetal life to about3yofageisthemost critical time period for gestational week. -
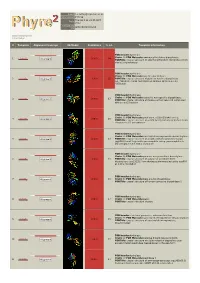
Phyre 2 Results for P15288
Email [email protected] Description P15288 Thu Jan 5 11:34:45 GMT Date 2012 Unique Job d2087d8d303c52c9 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:hydrolase Chain: B: PDB Molecule:aminoacyl-histidine dipeptidase; 1 c3mruB_ 100.0 64 Alignment PDBTitle: crystal structure of aminoacylhistidine dipeptidase from vibrio2 alginolyticus PDB header:hydrolase Chain: B: PDB Molecule:xaa-his dipeptidase; 2 c2qyvB_ Alignment 100.0 55 PDBTitle: crystal structure of putative xaa-his dipeptidase (yp_718209.1) from2 haemophilus somnus 129pt at 2.11 a resolution PDB header:hydrolase Chain: A: PDB Molecule:cytosolic non-specific dipeptidase; 3 c2zogA_ 100.0 17 Alignment PDBTitle: crystal structure of mouse carnosinase cn2 complexed with zn and2 bestatin PDB header:hydrolase Chain: B: PDB Molecule:peptidase, m20/m25/m40 family; 4 c2pokB_ 100.0 16 Alignment PDBTitle: crystal structure of a m20 family metallo peptidase from streptococcus2 pneumoniae PDB header:hydrolase Chain: A: PDB Molecule:succinyl-diaminopimelate desuccinylase; 5 c3pfeA_ Alignment 100.0 13 PDBTitle: crystal structure of a m20a metallo peptidase (dape, lpg0809) from2 legionella pneumophila subsp. pneumophila str. philadelphia 1 at 1.503 a resolution PDB header:hydrolase Chain: B: PDB Molecule:putative acetylornithine deacetylase; 6 c3pfoB_ Alignment 100.0 16 PDBTitle: crystal structure of a putative acetylornithine deacetylase (rpa2325)2 from rhodopseudomonas palustris cga009 at 1.90 a resolution PDB