Rnase PH: an Escherichia Coli Phosphate-Dependent Nuclease Distinct from Polynucleotide Phosphorylase (Trna Processing/Phosphorolysis) MURRAY P
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -

FUNCTIONAL and MECHANISTIC CHARACTERIZATION of TWO TRNA MODIFYING ENZYMES LAURA CAROLE KEFFER-WILKES Master of Science, Universi
FUNCTIONAL AND MECHANISTIC CHARACTERIZATION OF TWO TRNA MODIFYING ENZYMES LAURA CAROLE KEFFER-WILKES Master of Science, University of Lethbridge, 2012 A Thesis Submitted to the School of Graduate Studies Of the University of Lethbridge In Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY Department of Chemistry and Biochemistry University of Lethbridge LETHBRIDGE, ALBERTA, CANADA © Laura Keffer-Wilkes, 2016 FUNCTIONAL AND MECHANISTIC CHARACTERIZATION OF TWO TRNA MODIFYING ENZYMES LAURA CAROLE KEFFER-WILKES Date of Defence: June 28, 2016 Dr. Ute Wieden-Kothe Associate Professor Ph.D. Supervisor Dr. Tony Russell Assistant Professor Ph.D. Thesis Examination Committee Member Dr. Stacey Wetmore Professor Ph.D. Thesis Examination Committee Member Dr. Hans-Joachim Wieden Professor Ph.D. Thesis Examination Committee Member Dr. Eugene Mueller Professor Ph.D. External Examiner University of Louisville Louisville, Kentucky Dr. Elizabeth Schultz Associate Professor Ph.D. Internal External Examiner University of Lethbridge Dr. Michael Gerken Professor Ph.D. Chair, Thesis Examination Committee Abstract The formation of pseudouridine (Ѱ) and 5-methyluridine (m5U) in the T-arm of transfer RNAs (tRNAs) is near-universally conserved. These two modifications are formed in Escherichia coli by the pseudouridine synthase TruB and the S-adenosylmethionine- dependent methyltransferase TrmA, respectively. In this thesis, I investigate the function and mechanisms of these two tRNA modifying enzymes. First, in vitro and in vivo analysis of TruB reveals that this enzyme is acting as a tRNA chaperone which proves a long outstanding hypothesis. Secondly, characterization of ligand binding by TrmA shows that binding is cooperative and disruption of tRNA elbow region tertiary interactions by TrmA is essential for efficient tRNA binding and catalysis, leading to future analysis. -

A Protein Subunit of Human Rnase P, Rpp14, and Its Interacting Partner, OIP2, Have 335 Exoribonuclease Activity
A protein subunit of human RNase P, Rpp14, and its interacting partner, OIP2, have 335 exoribonuclease activity Taijiao Jiang and Sidney Altman* Department of Molecular, Cellular and Developmental Biology, Yale University, New Haven, CT 06520 Contributed by Sidney Altman, February 12, 2002 -The processing of precursor tRNAs at their 5 and 3 termini is a presence of [␣-32P]UTP. Internally labeled SupS1-3Ј was pre fundamental event in the biosynthesis of tRNA. RNase P is gener- pared from internally labeled 5Ј-SupS1-3Ј by removing the 5Ј ally responsible for endonucleolytic removal of a leader sequence leader sequence by using purified human RNase P. For the of precursor tRNA to generate the mature 5 terminus. However, synthesis of 5Ј-32P-labeled 5Ј-Tyr-3Ј, transcription reactions were much less is known about the RNase P counterparts or other performed with nonlabeled NTPs, the RNA was purified and proteins that are active at the tRNA 3 terminus. Here we show that then was treated with calf intestinal phosphatase, extracted two one of the human RNase P subunits, Rpp14, together with one of times with phenol͞chloroform and one time with chloroform. its interacting protein partners, OIP2, is a 335 exoribonuclease Recovery was by ethanol precipitation. 5Ј-Tyr-3Ј was labeled .with a phosphorolytic activity that processes the 3 terminus of with [␥-32P]ATP by using polynucleotide kinase precursor tRNA. Immunoprecipitates of a crude human RNase P Adult enhancer factor-1 (AEF) RNA, a single-stranded RNA complex can process both ends of precursor tRNA by hydrolysis, from Drosophila melanogaster (12), was labeled as indicated by but purified RNase P has no exonuclease activity. -

B Number Gene Name Strand Orientation Protein Length Mrna
list list sample) short list predicted B number Operon ID Gene name assignment Protein length mRNA present mRNA intensity Gene description Protein detected - Strand orientation Membrane protein detected (total list) detected (long list) membrane sample Proteins detected - detected (short list) # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides Functional category detected (membrane Protein detected - total Protein detected - long b0001 thrL + 21 1344 P 1 0 0 0 0 thr operon leader peptide Metabolism of small molecules 1 b0002 thrA + 820 13624 P 39 P 18 P 18 P 18 P(m) 2 aspartokinase I, homoserine dehydrogenase I Metabolism of small molecules 1 b0003 thrB + 310 6781 P 9 P 3 3 P 3 0 homoserine kinase Metabolism of small molecules 1 b0004 thrC + 428 15039 P 18 P 10 P 11 P 10 0 threonine synthase Metabolism of small molecules 1 b0005 b0005 + 98 432 A 5 0 0 0 0 orf, hypothetical protein Open reading frames 2 b0006 yaaA - 258 1047 P 11 P 1 2 P 1 0 orf, hypothetical protein Open reading frames 3 b0007 yaaJ - 476 342 P 8 0 0 0 0 MP-GenProt-PHD inner membrane transport protein Miscellaneous 4 b0008 talB + 317 20561 P 20 P 13 P 16 P 13 0 transaldolase B Metabolism of small molecules 5 b0009 mog + 195 1296 P 7 0 0 0 0 required for the efficient incorporation of molybdate into molybdoproteins Metabolism of small molecules 6 b0010 yaaH - 188 407 A 2 0 0 0 0 PHD orf, hypothetical protein Open reading frames 7 b0011 b0011 - 237 338 P 13 0 0 0 0 putative oxidoreductase Miscellaneous 8 b0012 htgA -

A Possible Functional Link Between RNA Degradation and Transcription in Bacillus Subtilis
A possible functional link between RNA degradation and transcription in Bacillus subtilis Dissertation for the award of the degree “Doctor of Philosophy” Division of Mathematics and Natural Sciences of the Georg-August-University Göttingen within the program “Microbiology and Biochemistry” of the Georg-August University School of Science (GAUSS) submitted by Martin Benda from Prague (Czech Republic) Göttingen 2020 Thesis Committee Prof. Dr. Jörg Stülke (Supervisor and 1st Reviewer) Institute for Microbiology and Genetics, Department of General Microbiology, Georg-August-University Göttingen Prof. Dr. Rolf Daniel (2nd Reviewer) Institute for Microbiology and Genetics, Department of Genomic and Applied Microbiology, Georg-August- University Göttingen Prof. Dr. Fabian M. Commichau Institute for Biotechnology, Department of Synthetic Microbiology, Brandenburg University of Technology Cottbus-Senftenberg Additional members of the Examination Board Prof. Dr. Markus Bohnsack Department of Molecular Biology, University Medical Center Göttingen Prof. Dr. Patrick Cramer Department of Molecular Biology, Max Planck Institute for Biophysical Chemistry, Göttingen Prof. Dr. Stefanie Pöggeler Institute for Microbiology and Genetics, Department of Genetics of Eukaryotic Microorganisms, Georg- August-University Göttingen Date of oral examination: September 17th, 2020 I hereby declare that this Ph.D. thesis entitled “A possible functional link between RNA degradation and transcription in Bacillus subtilis” has been written independently and with no other sources and aids than quoted. Martin Benda Acknowledgements First of all, I would like to express my deep thanks to my supervisor Prof. Dr. Jörg Stülke for the great guidance during this work. He put his full trust in me and gave me the opportunity to work on this intricate, but engaging topic of RNA degradation. -

An Investigation of Bacterial Ribonucleases As an Antibiotic Target Ashley Denise Frazier East Tennessee State University
East Tennessee State University Digital Commons @ East Tennessee State University Electronic Theses and Dissertations Student Works 5-2012 An Investigation of Bacterial Ribonucleases as an Antibiotic Target Ashley Denise Frazier East Tennessee State University Follow this and additional works at: https://dc.etsu.edu/etd Part of the Bacteriology Commons Recommended Citation Frazier, Ashley Denise, "An Investigation of Bacterial Ribonucleases as an Antibiotic Target" (2012). Electronic Theses and Dissertations. Paper 1417. https://dc.etsu.edu/etd/1417 This Dissertation - Open Access is brought to you for free and open access by the Student Works at Digital Commons @ East Tennessee State University. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons @ East Tennessee State University. For more information, please contact [email protected]. An Investigation of Bacterial Ribonucleases as an Antibiotic Target A dissertation presented to the faculty of the Department of Biochemistry and Molecular Biology East Tennessee State University In partial fulfillment of the requirements for the degree Doctor of Philosophy of Biomedical Sciences by Ashley Denise Frazier May 2012 W. Scott Champney, Ph.D., Chair Mitchell E. Robinson, Ph.D. Douglas P. Thewke, Ph.D. J. Russell Hayman, Ph.D. Bert C. Lampson, Ph.D. Keywords: Ribonuclease Mutants, Ribosomal Subunit Assembly, Aminoglycosides, Escherichia coli, Staphylococcus aureus, Protein Synthesis, Vanadyl Ribonucleoside Complex ABSTRACT An Investigation of Bacterial Ribonucleases as an Antibiotic Target by Ashley Denise Frazier Antibiotics have been commonly used in medical practice for over 40 years. However, the misuse and overuse of current antibiotics is thought to be the primary cause for the increase in antibiotic resistance. -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine -

Product Sheet Info
Master Clone List for NR-19280 Yersinia pestis Strain KIM Gateway® Clone Set, Recombinant in Escherichia coli, Plates 1-43 Catalog No. NR-19280 Table 1: Yersinia pestis Gateway® Clone, Plate 1 (UYPVA), NR-195971 Clone Well Locus ID Description ORF Accession Average Position Length Number Depth of Coverage 38250 A01 NTL02YP3655 transcriptional activator of nhaA 900 AAM87251.1 3.90531915 38268 A02 NTL02YP0999 apbA protein 912 AAM84595.1 6.01785714 38344 A03 NTL02YP3653 putative regulator 939 AAM87249.1 5.6680286 38375 A04 NTL02YP3668 homoserine kinase 951 AAM87264.1 5.57214934 38386 A05 NTL02YP1006 cytochrome o ubiquinol oxidase subunit II 957 AAM84602.1 5.45937813 38408 A06 NTL02YP2211 suppressor of htrB, heat shock protein 963 AAM85807.1 5.40877368 38447 A07 NTL02YP3649 penicillin tolerance protein 978 AAM87245.1 5.34086444 38477 A08 NTL02YP0988 thiamin-monophosphate kinase 990 AAM84584.1 6.8223301 36045 A09 NTL02YP3670 hypothetical protein 159 AAM87266.1 4.53266332 36094 A10 NTL02YP3672 hypothetical protein 174 AAM87268.1 4.72429907 36136 A11 NTL02YP1000 hypothetical protein 189 AAM84596.1 5.25327511 36241 A12 NTL02YP2577 hypothetical protein 225 AAM86173.1 3.88301887 membrane-bound ATP synthase, F0 36280 B01 NTL02YP4086 240 AAM87682.1 4.79285714 sector, subunit c 36342 B02 NTL02YP3654 30S ribosomal subunit protein S20 264 AAM87250.1 3.86513158 36362 B03 NTL02YP2570 hypothetical protein 270 AAM86166.1 5.40322581 38504 B04 NTL02YP2573 putative ABC transporter permease 996 AAM86169.1 6.65444015 38516 B05 NTL02YP0234 putative heat shock -

Synthesis of RNA with Selective Isotopic Labels for NMR Structural Studies
Synthesis of RNA with Selective Isotopic Labels for NMR Structural Studies by Thomas J. Tolbert B.S., Chemistry Purdue University, 1991 Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY IN BIOCHEMISTRY at the Massachusetts Institute of Technology February 1998 @ Massachusetts Institute of Technology, 1998 All Rights Reserved Signature of Author ...... ,..., .. ..... ....... ..................... SDepartment of Chemistry November 19, 1997 CCertified ertified bby......................W.... y ...................... ..... ....... I..................... ............................ James R. Williamson Associate Professor of Chemistry Thesis Supervisor Accepted by............. Dietmar Seyferth Chair, Departmental Committee on Graduate Students This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: Professor JoAnne Stubbe....... ....................... ..... C hair Chair Professor James R. Williamson.............................. ............ ,-7 Thesis Supervisor Professor Lawrence J. Stern.. ........... .. .......... Synthesis of RNA with Selective Isotopic Labels for NMR Structural Studies by Thomas J. Tolbert Submitted to the Department of Chemistry on November 19, 1997 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biochemistry ABSTRACT RNA is synthesized with specific isotopic labels (2 H, 13 C, and 15N) to alleviate spectral crowding and broad linewidth problems encountered in large RNA NMR spectroscopy. A combined chemical and enzymatic synthesis is described in which glycerol (U-d8) is chemically converted into 3',4',5',5"- 2 H4-D,L-ribose, and then the 3',4',5',5"- 2 H4-D-ribose is enzymatically converted into the four NTPs of RNA. Enzymes of purine salvage and pyrimidine biosynthesis are overexpressed and purified in order to catalyze the nucleotide forming reactions. Several isotopically labeled forms of glucose are enzymatically converted into the four NTPs of RNA. -
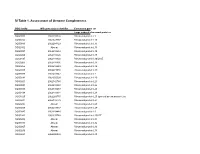
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Crystal Structure of Escherichia Coli Polynucleotide Phosphorylase Core Bound to Rnase E, RNA and Manganese: Implications for Ca
doi:10.1016/j.jmb.2009.03.051 J. Mol. Biol. (2009) 389,17–33 Available online at www.sciencedirect.com Crystal Structure of Escherichia coli Polynucleotide Phosphorylase Core Bound to RNase E, RNA and Manganese: Implications for Catalytic Mechanism and RNA Degradosome Assembly Salima Nurmohamed1, Bhamini Vaidialingam1, Anastasia J. Callaghan2 and Ben F. Luisi1⁎ 1Department of Biochemistry, Polynucleotide phosphorylase (PNPase) is a processive exoribonuclease University of Cambridge, that contributes to messenger RNA turnover and quality control of ribo- 80 Tennis Court Road, somal RNA precursors in many bacterial species. In Escherichia coli, a pro- Cambridge CB2 1GA, UK portion of the PNPase is recruited into a multi-enzyme assembly, known as the RNA degradosome, through an interaction with the scaffolding do- 2Institute of Biomedical and main of the endoribonuclease RNase E. Here, we report crystal structures of Biomolecular Sciences, E. coli PNPase complexed with the recognition site from RNase E and University of Portsmouth, with manganese in the presence or in the absence of modified RNA. The Portsmouth, PO1 2DY, UK homotrimeric PNPase engages RNase E on the periphery of its ring-like Received 31 October 2008; architecture through a pseudo-continuous anti-parallel β-sheet. A similar received in revised form interaction pattern occurs in the structurally homologous human exosome 5 March 2009; between the Rrp45 and Rrp46 subunits. At the centre of the PNPase ring is a accepted 11 March 2009 tapered channel with an adjustable aperture where RNA bases stack on Available online phenylalanine side chains and trigger structural changes that propagate to 24 March 2009 the active sites. -

Structural Characterization of Proteinaceous Rnase P from Arabidopsis Thaliana Franziska Pinker
Structural characterization of proteinaceous RNase P from Arabidopsis thaliana Franziska Pinker To cite this version: Franziska Pinker. Structural characterization of proteinaceous RNase P from Arabidopsis thaliana. Biologie végétale. Université de Strasbourg, 2014. Français. NNT : 2014STRAJ093. tel-01236246 HAL Id: tel-01236246 https://tel.archives-ouvertes.fr/tel-01236246 Submitted on 1 Dec 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. UNIVERSITÉ DE STRASBOURG ÉCOLE DOCTORALE des Sciences de la Vie et de la Santé Institut de Biologie Moléculaire des Plantes - IBMP THÈSE présentée par : Franziska PINKER soutenue le : 15 septembre 2014 pour obtenir le grade de : !"#$%&'($')R%+ iversité de Strasbourg Discipline/ Spécialité : Sciences du Vivant /Biochimie, Biologie Moléculaire et Structurale Structural characterization of proteinaceous RNase P from Arabidopsis thaliana THÈSE dirigée par : M. Claude SAUTER Dr, Université de Strasbourg M. Philippe GIEGE Dr, Université de Strasbourg RAPPORTEURS : M. Walter ROSSMANITH Dr, Université de Vienne Mme Emmanuelle SCHMITT Dr, Ecole Polytechnique AUTRES MEMBRES DU JURY : Mme Anita MARCHFELDER Professeur, !"#$%&"' )*R ,- Mme Pascale ROMBY Dr, Université de Strasbourg Fur¨ meine Familie Acknowledgements First of all I would like to thank the members of my jury, Dr.