Phase Behaviour in Water/Hydrocarbon Mixtures Involved in Gas Production Systems Antonin Chapoy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Midas Arsine
® Midas SENSOR CARTRIDGE SPECIFICATIONS Arsine (AsH3), Germane (GeH4) MIDAS-S-ASH, MIDAS-E-ASH Gas Measured Measured Arsine (AsH3) Cross Sensitivities Each Midas® sensor is potentially cross sensitive to other gases Cartridge Part Number MIDAS-S-ASH 1 year standard warranty MIDAS-E-ASH 2 year extended warranty and this may cause a gas reading when exposed to other gases than those originally designated. The table below presents typical Sensor Technology 3 electrode electrochemical cell readings that will be observed when a new sensor cartridge is Measuring Range (ppm) AsH 0 – 0.2ppm exposed to the cross sensitive gas (or a mixture of gases containing 3 the cross sensitive species). Minimum Alarm 1 Set Point 0.025ppm Gas / Vapor Chemical Concentration Reading Repeatability < ± 2% of measured value Formula applied (ppm) (ppm AsH3) Linearity < ± 10% of measured value Ammonia NH3 108 <0.1 Response Time t62.5 < 15 seconds Carbon Dioxide CO2 5,000 0 Sensor Cartridge Life Expectancy ≥ 24 months under typical application conditions Carbon Monoxide CO 85 0 Operating Temperature 0°C to + 40°C (32°C to 104°F) Chlorine CI2 TBD <-0.05 Effect of Temperature Diborane B2H6 0.1 0.05 Zero < ± 0.004 ppm / °C (0° to 40°C) Sensitivity < ± 0.9% of measured value / °C Disilane Si2H6 0.27 0.12 Operating Humidity (continuous) 10 – 95% rH non-condensing Germane GeH4 0.27 0.05 Effect of Humidity Hydrogen H2 3100 <-0.05 Zero Initial short term drift at abrupt rH change (< 0.001 ppm / % rH) Sensitivity < ± 0.2% of measured value / % rH Hydrogen Chloride HCI 7.9 0 -

Flow Injection Electrochemical Hydride Generation of Hydrogen Selenide on Lead Cathode: Critical Study of the Influence of Experimental Parameters
ANALYTICAL SCIENCES MARCH 2003, VOL. 19 367 2003 © The Japan Society for Analytical Chemistry Flow Injection Electrochemical Hydride Generation of Hydrogen Selenide on Lead Cathode: Critical Study of the Influence of Experimental Parameters Eduardo BOLEA, Francisco LABORDA,† and Juan R. CASTILLO Analytical Spectroscopy and Sensors Group, Department of Analytical Chemistry, University of Zaragoza, Zaragoza, Spain A flow injection system for the determination of selenium by electrochemical hydride generation and quartz tube atomic absorption spectrometry is described. The generator consists of an electrolytic flow-through cell with a concentric arrangement and a packed cathode made of particulated lead. The influences of sample flow rate, carrier gas flow rate and electrolysis current on the hydrogen selenide generation have been critically studied. Both sample flow rate and electrolysis current play important roles in the efficiency of the hydride generation process. A characteristic mass of 2.4 ng and a concentration detection limit of 17 µg l–1 were obtained for a sample volume of 420 µl. (Received January 22, 2002; Accepted September 25, 2002) The electrochemical generation of different hydrides on lead Introduction cathodes, the material that offers the highest hydrogen overpotential among those commonly used, has been studied by The formation of volatile covalent hydrides of a number of several authors.3–5,7–11 Sand3 and Thompson4,5 obtained elements (As, Bi, Ge, Sn, Sb, Se, Te and Pb) by reaction with generation efficiencies of 100%, whereas efficiencies of 86,11 sodium tetrahydroborate in acidic media has been widely used 928 and 60%11 were obtained for arsine, stibine and hydrogen for sample introduction in atomic spectrometry. -

Biological Chemistry of Hydrogen Selenide
antioxidants Review Biological Chemistry of Hydrogen Selenide Kellye A. Cupp-Sutton † and Michael T. Ashby *,† Department of Chemistry and Biochemistry, University of Oklahoma, Norman, OK 73019, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-405-325-2924 † These authors contributed equally to this work. Academic Editors: Claus Jacob and Gregory Ian Giles Received: 18 October 2016; Accepted: 8 November 2016; Published: 22 November 2016 Abstract: There are no two main-group elements that exhibit more similar physical and chemical properties than sulfur and selenium. Nonetheless, Nature has deemed both essential for life and has found a way to exploit the subtle unique properties of selenium to include it in biochemistry despite its congener sulfur being 10,000 times more abundant. Selenium is more easily oxidized and it is kinetically more labile, so all selenium compounds could be considered to be “Reactive Selenium Compounds” relative to their sulfur analogues. What is furthermore remarkable is that one of the most reactive forms of selenium, hydrogen selenide (HSe− at physiologic pH), is proposed to be the starting point for the biosynthesis of selenium-containing molecules. This review contrasts the chemical properties of sulfur and selenium and critically assesses the role of hydrogen selenide in biological chemistry. Keywords: biological reactive selenium species; hydrogen selenide; selenocysteine; selenomethionine; selenosugars; selenophosphate; selenocyanate; selenophosphate synthetase thioredoxin reductase 1. Overview of Chalcogens in Biology Chalcogens are the chemical elements in group 16 of the periodic table. This group, which is also known as the oxygen family, consists of the elements oxygen (O), sulfur (S), selenium (Se), tellurium (Te), and the radioactive element polonium (Po). -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

2020 Emergency Response Guidebook
2020 A guidebook intended for use by first responders A guidebook intended for use by first responders during the initial phase of a transportation incident during the initial phase of a transportation incident involving hazardous materials/dangerous goods involving hazardous materials/dangerous goods EMERGENCY RESPONSE GUIDEBOOK THIS DOCUMENT SHOULD NOT BE USED TO DETERMINE COMPLIANCE WITH THE HAZARDOUS MATERIALS/ DANGEROUS GOODS REGULATIONS OR 2020 TO CREATE WORKER SAFETY DOCUMENTS EMERGENCY RESPONSE FOR SPECIFIC CHEMICALS GUIDEBOOK NOT FOR SALE This document is intended for distribution free of charge to Public Safety Organizations by the US Department of Transportation and Transport Canada. This copy may not be resold by commercial distributors. https://www.phmsa.dot.gov/hazmat https://www.tc.gc.ca/TDG http://www.sct.gob.mx SHIPPING PAPERS (DOCUMENTS) 24-HOUR EMERGENCY RESPONSE TELEPHONE NUMBERS For the purpose of this guidebook, shipping documents and shipping papers are synonymous. CANADA Shipping papers provide vital information regarding the hazardous materials/dangerous goods to 1. CANUTEC initiate protective actions. A consolidated version of the information found on shipping papers may 1-888-CANUTEC (226-8832) or 613-996-6666 * be found as follows: *666 (STAR 666) cellular (in Canada only) • Road – kept in the cab of a motor vehicle • Rail – kept in possession of a crew member UNITED STATES • Aviation – kept in possession of the pilot or aircraft employees • Marine – kept in a holder on the bridge of a vessel 1. CHEMTREC 1-800-424-9300 Information provided: (in the U.S., Canada and the U.S. Virgin Islands) • 4-digit identification number, UN or NA (go to yellow pages) For calls originating elsewhere: 703-527-3887 * • Proper shipping name (go to blue pages) • Hazard class or division number of material 2. -

3 • Molecules and Compounds
South Pasadena · AP Chemistry Name ____________________________________ Period ___ Date ___/___/___ 3 · Molecules and Compounds STUDY QUESTIONS and PROBLEMS 1. a. The structural formula for acetic acid is CH3CO2H. What is its empirical formula; what is its molecular formula? b. The molecular formula of acrylonitrile is C3H3N. Look up in the text, and draw, its structural formula. c. The molecular formula of aspartame (nutrasweet) is C14H18O5N2. Look up in the text, and draw, its structural formula. 2. The formulas for ethanol and ammonium nitrate are C2H5OH and NH4NO3. In what respects are these formulas and compounds different? 3. The molecular formula for both butanol and diethylether is C4H10O. Write structural formulas for both and show how they are different. Are any other structures possible? 4. Name the polyatomic ions: - - CH3CO2 HCO3 - 2- H2PO4 Cr2O7 2- - SO3 ClO 4 5. What are the formulas of the polyatomic ions: phosphate nitrite sulfate cyanide bisulfite chlorite 6. Write the ions present in the following salts and predict their formulas: potassium bromide calcium carbonate magnesium iodide lithium oxide aluminum sulfate ammonium chlorate beryllium phosphate 7. Name the following ionic salts: (NH4)2SO4 Co2(SO4)3 KHCO3 NiSO4 Ca(NO3)2 AlPO4 8. Name the following binary compounds of the nonmetals: CS2 SiCl4 SF6 GeH4 IF5 P4O10 N2H4 S4N4 PCl5 OF2 Cl2O7 IF7 9. What are the formulas for the following binary compounds? silicon dioxide phosphine boron trifluoride silicon carbide xenon tetroxide phosphorus tribromide dinitrogen pentoxide disulfur dichloride bromine trifluroide hydrogen selenide carbon tetrachloride 10. a. How many moles are present in 128 grams of sulfur dioxide? b. -
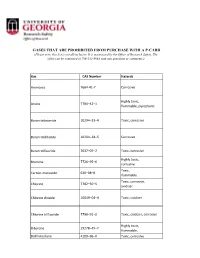
Lists of Gases for Pcard Manual.Xlsx
GASES THAT ARE PROHIBITED FROM PURCHASE WITH A P-CARD (Please note, this list is not all-inclusive. It is maintained by the Office of Research Safety. The office can be contacted at 706-542-9088 with any questions or comments.) Gas CAS Number Hazards Ammonia 7664‐41‐7 Corrosive Highly toxic, Arsine 7784–42–1 flammable, pyrophoric Boron tribromide 10294–33–4 Toxic, corrosive Boron trichloride 10294–34–5 Corrosive Boron trifluoride 7637–07–2 Toxic, corrosive Highly toxic, Bromine 7726–95–6 corrosive Toxic, Carbon monoxide 630–08–0 flammable Toxic, corrosive, Chlorine 7782–50–5 oxidizer Chlorine dioxide 10049–04–4 Toxic, oxidizer Chlorine trifluoride 7790–91–2 Toxic, oxidizer, corrosive Highly toxic, Diborane 19278–45–7 flammable, Dichlorosilane 4109–96–0 Toxic, corrosive Ethylene oxide 75–21–8 Toxic, flammable Highly toxic, corrosive, Fluorine 7782–41–4 oxidizer Highly toxic, Germane 7782–65–2 flammable Hydrogen bromide 10035–10–6 Toxic, corrosive Hydrogen chloride 7647–01–0 Toxic, corrosive Hydrogen cyanide 74–90–8 Highly toxic, flammable Hydrogen fluoride 7664–39–3 Toxic, corrosive Hydrogen iodide 10034‐85‐2 Toxic, corrosive Highly toxic, Hydrogen selenide 7783–07–5 flammable Toxic, flammable, Hydrogen sulfide 7783–06–4 corrosive Methyl bromide 74–83–9 Toxic Methyl isocyanate 624‐83‐9 Highly toxic, flammable Methyl mercaptan 74–93–1 Toxic, flammable Nickel carbonyl 13463–39–3 Highly toxic, flammable Highly toxic, Nitric oxide 10102–43–9 oxidizer Highly toxic, Nitrogen dioxide 10102–44–0 oxidizer, corrosive Highly toxic, Ozone 10028‐15‐6 -

Safetygram 34: the Toxic Metal Hydrides
Safetygram #34 The Toxic Metal Hydrides Arsine, Diborane, Germane, Hydrogen Selenide, Phosphine General These products are members of the compound to 17237 kPa). In addition to their extreme toxic- family called metal hydrides. Hydrides are com- ity, these products are also highly flammable. pounds of hydrogen with a more electropositive Toxic metal hydrides are commonly used in the element. Arsine, diborane, germane, hydrogen manufacture of semiconductors, as they are used selenide, and phosphine are members of a subset to deposit the base element into the structures of family called toxic metal hydrides because of their semiconductor circuits to change the properties or significant toxicity. The scope of this Safetygram grow crystals. will be limited to these five products, which are all liquefied compressed gases. Liquefied compressed The highly toxic nature of these products has moti- gases are gases that, when compressed in a con- vated many regulatory and code organizations to tainer, partially liquefy at ordinary temperatures develop strict rules for the storage and use of these and pressures ranging from 25 to 2500 psig (172 products. Because of their hazardous properties, the purchase of these products is controlled. Table 1 Physical and Chemical Properties Property Arsine Diborane Germane Hydrogen Phosphine Selenide Molecular Formula AsH3 B2H6 GeH4 H2Se PH3 Molecular Weight 77.95 27.67 76.62 80.98 34.0 Specific Gravity (air=1) 2.691 0.96 2.66 2.12 1.174 Vapor Pressure @ 70ºF psia 217.9 536.55* 640 137.78 493.2 @ 21.1ºC kPa, abs 1502 3699* 4413 950 3400 Boiling Point ºF -79.9 -134.8 -127.3 -42 -126 ºC -62 -93 -88.5 -41 -88 Specific Volume (scf/lb) 4.91 13.86 5.05 4.8 11.3 (m3/kg) 0.306 0.865 0.315 0.299 0.705 Gas Density (lb/scf) 0.204 0.072 0.2 0.209 0.088 (kg/m3) 3.268 1.153 3.204 3.348 1.41 *vapor pressure is at 60ºF (15.6ºC) Warning: Improper storage, handling, or use of toxic metal hydrides can result in serious injury and/or property damage. -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

This Table Gives the Standard State Chemical Thermodynamic Properties of About 2500 Individual Substances in the Crystalline, Liquid, and Gaseous States
STANDARD THERMODYNAMIC PROPERTIES OF CHEMICAL SUBSTANCES This table gives the standard state chemical thermodynamic properties of about 2500 individual substances in the crystalline, liquid, and gaseous states. Substances are listed by molecular formula in a modified Hill order; all substances not containing carbon appear first, followed by those that contain carbon. The properties tabulated are: DfH° Standard molar enthalpy (heat) of formation at 298.15 K in kJ/mol DfG° Standard molar Gibbs energy of formation at 298.15 K in kJ/mol S° Standard molar entropy at 298.15 K in J/mol K Cp Molar heat capacity at constant pressure at 298.15 K in J/mol K The standard state pressure is 100 kPa (1 bar). The standard states are defined for different phases by: • The standard state of a pure gaseous substance is that of the substance as a (hypothetical) ideal gas at the standard state pressure. • The standard state of a pure liquid substance is that of the liquid under the standard state pressure. • The standard state of a pure crystalline substance is that of the crystalline substance under the standard state pressure. An entry of 0.0 for DfH° for an element indicates the reference state of that element. See References 1 and 2 for further information on reference states. A blank means no value is available. The data are derived from the sources listed in the references, from other papers appearing in the Journal of Physical and Chemical Reference Data, and from the primary research literature. We are indebted to M. V. Korobov for providing data on fullerene compounds. -

Midas Diborane
® Midas SENSOR CARTRIDGE SPECIFICATIONS Diborane (B2H6) MIDAS-S-B2H, MIDAS-E-B2H Gas Measured Diborane B H Cross Sensitivities 2 6 Each Midas® sensor is potentially cross sensitive to other gases and Cartridge Part Number MIDAS-S-B2H 1 year standard warranty this may cause a gas reading when exposed to other gases than MIDAS-E-B2H 2 year extended warranty those originally designated. The table below presents typical readings Sensor Technology 3 electrode electrochemical cell that will be observed when a new sensor cartridge is exposed to the cross sensitive gas (or a mixture of gases containing the cross Measuring Range (ppm) B H 0 – 0.4ppm 2 6 sensitive species). Minimum Alarm 1 Set Point 0.050ppm Gas / Vapor Chemical Concentration Reading Repeatability < ± 2% of measured value Formula applied (ppm) (ppm B2H6) Linearity < ± 10% of Full Scale Ammonia NH3 108 <0.1 Arsine AsH 0.2 0.1 Response Time t62.5 < 15 seconds 3 Sensor Cartridge Life Expectancy ≥ 24 months under typical application conditions Carbon Dioxide CO2 5,000 0 Operating Temperature 0°C to +40°C (32°F to 104°F) Carbon Monoxide CO 85 0 Effect of Temperature Zero: < ± 0.0002ppm / °C (0°C to 40°C) Chlorine Cl 0.85 -0.15 Sensitivity: < ± 1% of measured value / °C 2 Disilane Si H 0.27 0.12 Operating Humidity (continuous) 10 – 95% rH (non-condensing) 2 6 Effect of Humidity Zero: Initial short term drift at abrupt RH change Germane GeH4 0.94 0.1 (< 0.0075ppm / % rH) Hydrogen H 3100 0 Sensitivity: < ± 0.5% of measured value / % rH 2 Operating Pressure 90 – 110kPa Hydrogen Chloride -

Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005)
NOMENCLATURE OF INORGANIC CHEMISTRY IUPAC Recommendations 2005 IUPAC Periodic Table of the Elements 118 1 2 21314151617 H He 3 4 5 6 7 8 9 10 Li Be B C N O F Ne 11 12 13 14 15 16 17 18 3456 78910 11 12 Na Mg Al Si P S Cl Ar 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe 55 56 * 57− 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 Cs Ba lanthanoids Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn 87 88 ‡ 89− 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 Fr Ra actinoids Rf Db Sg Bh Hs Mt Ds Rg Uub Uut Uuq Uup Uuh Uus Uuo * 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu ‡ 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr International Union of Pure and Applied Chemistry Nomenclature of Inorganic Chemistry IUPAC RECOMMENDATIONS 2005 Issued by the Division of Chemical Nomenclature and Structure Representation in collaboration with the Division of Inorganic Chemistry Prepared for publication by Neil G.