A Mechanistic Investigation of Insulin Receptor Substrate 2 Function in Breast Cancer Progression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
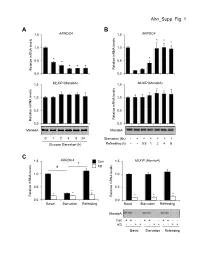
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

Transcriptomic Characterisation of the Molecular Mechanisms Induced by Rgma During Skeletal Muscle Hyperplasia and Hypertrophy
Transcriptomic Characterisation of the Molecular Mechanisms Induced by RGMa During Skeletal Muscle Hyperplasia and Hypertrophy Aline Gonçalves Lio Copola Universidade Federal de Minas Gerais Íria Gabriela Dias dos Santos Universidade Federal de Minas Gerais Luiz Lehmann Coutinho Universidade de São Paulo Luiz Eduardo Vieira Del Bem Universidade Federal de Minas Gerais Paulo Henrique de Almeida Campos Junior Federal University of São João del-Rei Júlia Meireles Nogueira Universidade Federal de Minas Gerais Alinne do Carmo Costa Universidade Federal de Minas Gerais Gerluza Aparecida Borges Silva Universidade Federal de Minas Gerais Erika Cristina Jorge ( [email protected] ) Universidade Federal de Minas Gerais Research Article Keywords: Axon Guidance, myogenesis, hypertrophy, hyperplasia, skeletal muscle differentiation, transcriptomic analysis Posted Date: June 29th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-646954/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/25 Abstract Background: The repulsive guidance molecule a (RGMa) is a GPI-anchor axon guidance molecule rst found to play important roles during neuronal development. RGMa expression patterns and signalling pathways via Neogenin and/or as BMP coreceptors indicated that this axon guidance molecule could also be working in other processes and diseases, including during myogenesis. Previous works have consistently shown that RGMa is expressed in skeletal muscle cells and that its overexpression induces both nuclei accretion and hypertrophy in muscle cell lineages. However, the cellular components and molecular mechanisms induced by RGMa during the differentiation of skeletal muscle cells are poorly understood. In this work, the global transcription expression prole of RGMa-treated C2C12 myoblasts during the differentiation stage, obtained by RNA- seq, were reported. -

Potential Applications for Mrna and Peptide-Based Vaccines
viruses Article An Epitope Platform for Safe and Effective HTLV-1-Immunization: Potential Applications for mRNA and Peptide-Based Vaccines Guglielmo Lucchese 1,*,†, Hamid Reza Jahantigh 2,3,† , Leonarda De Benedictis 2, Piero Lovreglio 2 and Angela Stufano 2,3 1 Department of Neurology, Medical University of Greifswald, 17475 Greifswald, Germany 2 Interdisciplinary Department of Medicine-Section of Occupational Medicine, University of Bari, 70124 Bari, Italy; [email protected] (H.R.J.); [email protected] (L.D.B.); [email protected] (P.L.); [email protected] (A.S.) 3 Animal Health and Zoonosis Doctoral Program, Department of Veterinary Medicine, University of Bari, 70010 Bari, Italy * Correspondence: [email protected] † These authors contributed equally to the work. Abstract: Human T-cell lymphotropic virus type 1 (HTLV-1) infection affects millions of individuals worldwide and can lead to severe leukemia, myelopathy/tropical spastic paraparesis, and numerous other disorders. Pursuing a safe and effective immunotherapeutic approach, we compared the viral polyprotein and the human proteome with a sliding window approach in order to identify oligopeptide sequences unique to the virus. The immunological relevance of the viral unique oligopeptides was assessed by searching them in the immune epitope database (IEDB). We found that Citation: Lucchese, G.; Jahantigh, HTLV-1 has 15 peptide stretches each consisting of uniquely viral non-human pentapeptides which H.R.; De Benedictis, L.; Lovreglio, P.; Stufano, A. An Epitope Platform for are ideal candidate for a safe and effective anti-HTLV-1 vaccine. Indeed, experimentally validated Safe and Effective HTLV-1 epitopes, as retrieved from the IEDB, contain peptide sequences also present in a vast number HTLV-1-Immunization: Potential of human proteins, thus potentially instituting the basis for cross-reactions. -

ONCOGENOMICS Mammary Luminal Epithelial Cells
Oncogene (2003) 22, 2680–2688 & 2003 Nature Publishing Group All rights reserved 0950-9232/03 $25.00 www.nature.com/onc cDNA microarray analysis of genes associated with ERBB2 (HER2/neu) overexpression in human mammary luminal epithelial cells Alan Mackay*,1,6, Chris Jones2,6, Tim Dexter2, Ricardo LA Silva3, Karen Bulmer2, Allison Jones2, Peter Simpson2, Robert A Harris1, Parmjit S Jat4, A Munro Neville1, Luiz FL Reis3, Sunil R Lakhani2,5 and Michael J O’Hare1 1LICR/UCL Breast Cancer Laboratory, University College London, London, UK; 2The Breakthrough Toby Robins Breast Cancer Research Centre, Institute of Cancer Research, London, UK; 3Ludwig Institute for Cancer Research, Sao Paolo, Brazil; 4Ludwig Institute for Cancer Research, University College London, London, UK; 5Royal Marsden Hospital, London, UK To investigate changes in gene expression associated with the disease within their lifetime. Overexpression of the ERBB2, expression profiling of immortalized human proto-oncogene ERBB2 (HER2/neu) is observed in 25– mammary luminal epithelial cells and variants expressing 30% of all such cancers, and is an established adverse a moderate and high level of ERBB2 has been carried out prognostic factor (Slamon et al., 1987, 1989; Ross and using cDNA microarrays corresponding to approximately Fletcher, 1999; Menard et al., 2001) yielding a median 6000 unique genes/ESTs. A total of 61 significantly up- or survival of 3years, compared with 6–7 years when downregulated (>2.0-fold) genes were identified and unassociated with ERBB2. Overexpression also corre- further validated by RT–PCR analysis as well as lates with tumor size, lymph node metastases, high microarray comparisons with a spontaneously ERBB2- nuclear grade, high percentage of S-phase cells, aneu- overexpressing breast cancer cell line and ERBB2-positive ploidy and estrogen receptor (ER) and progesterone primary breast tumors. -

Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
www.nature.com/scientificreports OPEN Assessment of a complete and classifed platelet proteome from genome‑wide transcripts of human platelets and megakaryocytes covering platelet functions Jingnan Huang1,2*, Frauke Swieringa1,2,9, Fiorella A. Solari2,9, Isabella Provenzale1, Luigi Grassi3, Ilaria De Simone1, Constance C. F. M. J. Baaten1,4, Rachel Cavill5, Albert Sickmann2,6,7,9, Mattia Frontini3,8,9 & Johan W. M. Heemskerk1,9* Novel platelet and megakaryocyte transcriptome analysis allows prediction of the full or theoretical proteome of a representative human platelet. Here, we integrated the established platelet proteomes from six cohorts of healthy subjects, encompassing 5.2 k proteins, with two novel genome‑wide transcriptomes (57.8 k mRNAs). For 14.8 k protein‑coding transcripts, we assigned the proteins to 21 UniProt‑based classes, based on their preferential intracellular localization and presumed function. This classifed transcriptome‑proteome profle of platelets revealed: (i) Absence of 37.2 k genome‑ wide transcripts. (ii) High quantitative similarity of platelet and megakaryocyte transcriptomes (R = 0.75) for 14.8 k protein‑coding genes, but not for 3.8 k RNA genes or 1.9 k pseudogenes (R = 0.43–0.54), suggesting redistribution of mRNAs upon platelet shedding from megakaryocytes. (iii) Copy numbers of 3.5 k proteins that were restricted in size by the corresponding transcript levels (iv) Near complete coverage of identifed proteins in the relevant transcriptome (log2fpkm > 0.20) except for plasma‑derived secretory proteins, pointing to adhesion and uptake of such proteins. (v) Underrepresentation in the identifed proteome of nuclear‑related, membrane and signaling proteins, as well proteins with low‑level transcripts. -

Minus End–Directed Motor KIFC3 Suppresses E-Cadherin Degradation by Recruiting USP47 to Adherens Junctions
M BoC | ARTICLE Minus end–directed motor KIFC3 suppresses E-cadherin degradation by recruiting USP47 to adherens junctions Kyoko Sako-Kubotaa, Nobutoshi Tanakaa, Shigenori Nagaea, Wenxiang Menga,b, and Masatoshi Takeichia aRIKEN Center for Developmental Biology, Kobe 650-0047, Japan; bState Key Laboratory of Molecular Developmental Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China ABSTRACT The adherens junction (AJ) plays a crucial role in maintaining cell–cell adhesion in Monitoring Editor epithelial tissues. Previous studies show that KIFC3, a minus end–directed kinesin motor, Kozo Kaibuchi moves into AJs via microtubules that grow from clusters of CAMSAP3 (also known as Nezha), Nagoya University a protein that binds microtubule minus ends. The function of junction-associated KIFC3, how- Received: Jul 30, 2014 ever, remains to be elucidated. Here we find that KIFC3 binds the ubiquitin-specific protease Revised: Sep 17, 2014 USP47, a protease that removes ubiquitin chains from substrates and hence inhibits protea- Accepted: Sep 18, 2014 some-mediated proteolysis, and recruits it to AJs. Depletion of KIFC3 or USP47 promotes cleavage of E-cadherin at a juxtamembrane region of the cytoplasmic domain, resulting in the production of a 90-kDa fragment and the internalization of E-cadherin. This cleavage de- pends on the E3 ubiquitin protein ligase Hakai and is inhibited by proteasome inhibitors. E- cadherin ubiquitination consistently increases after depletion of KIFC3 or USP47. These find- ings suggest that KIFC3 suppresses the ubiquitination and resultant degradation of E-cadherin by recruiting USP47 to AJs, a process that may be involved in maintaining stable cell–cell adhesion in epithelial sheets. -

The Pdx1 Bound Swi/Snf Chromatin Remodeling Complex Regulates Pancreatic Progenitor Cell Proliferation and Mature Islet Β Cell
Page 1 of 125 Diabetes The Pdx1 bound Swi/Snf chromatin remodeling complex regulates pancreatic progenitor cell proliferation and mature islet β cell function Jason M. Spaeth1,2, Jin-Hua Liu1, Daniel Peters3, Min Guo1, Anna B. Osipovich1, Fardin Mohammadi3, Nilotpal Roy4, Anil Bhushan4, Mark A. Magnuson1, Matthias Hebrok4, Christopher V. E. Wright3, Roland Stein1,5 1 Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 2 Present address: Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN 3 Department of Cell and Developmental Biology, Vanderbilt University, Nashville, TN 4 Diabetes Center, Department of Medicine, UCSF, San Francisco, California 5 Corresponding author: [email protected]; (615)322-7026 1 Diabetes Publish Ahead of Print, published online June 14, 2019 Diabetes Page 2 of 125 Abstract Transcription factors positively and/or negatively impact gene expression by recruiting coregulatory factors, which interact through protein-protein binding. Here we demonstrate that mouse pancreas size and islet β cell function are controlled by the ATP-dependent Swi/Snf chromatin remodeling coregulatory complex that physically associates with Pdx1, a diabetes- linked transcription factor essential to pancreatic morphogenesis and adult islet-cell function and maintenance. Early embryonic deletion of just the Swi/Snf Brg1 ATPase subunit reduced multipotent pancreatic progenitor cell proliferation and resulted in pancreas hypoplasia. In contrast, removal of both Swi/Snf ATPase subunits, Brg1 and Brm, was necessary to compromise adult islet β cell activity, which included whole animal glucose intolerance, hyperglycemia and impaired insulin secretion. Notably, lineage-tracing analysis revealed Swi/Snf-deficient β cells lost the ability to produce the mRNAs for insulin and other key metabolic genes without effecting the expression of many essential islet-enriched transcription factors. -

Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared Amongst Δγ T, Innate Lymphoid, and Th Cells
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 δγ is online at: average * The Journal of Immunology , 10 of which you can access for free at: 2016; 197:1460-1470; Prepublished online 6 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600643 http://www.jimmunol.org/content/197/4/1460 Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst T, Innate Lymphoid, and Th Cells You Jeong Lee, Gabriel J. Starrett, Seungeun Thera Lee, Rendong Yang, Christine M. Henzler, Stephen C. Jameson and Kristin A. Hogquist J Immunol cites 41 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2016/07/06/jimmunol.160064 3.DCSupplemental This article http://www.jimmunol.org/content/197/4/1460.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 26, 2021. The Journal of Immunology Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst gd T, Innate Lymphoid, and Th Cells You Jeong Lee,* Gabriel J. -

Murine Perinatal Beta Cell Proliferation and the Differentiation of Human Stem Cell Derived Insulin Expressing Cells Require NEUROD1
Page 1 of 105 Diabetes Murine perinatal beta cell proliferation and the differentiation of human stem cell derived insulin expressing cells require NEUROD1 Anthony I. Romer,1,2 Ruth A. Singer1,3, Lina Sui2, Dieter Egli,2* and Lori Sussel1,4* 1Department of Genetics and Development, Columbia University, New York, NY 10032, USA 2Department of Pediatrics, Columbia University, New York, NY 10032, USA 3Integrated Program in Cellular, Molecular and Biomedical Studies, Columbia University, New York, NY 10032, USA 4Department of Pediatrics, University of Colorado Denver School of Medicine, Denver, CO 80045, USA *Co-Corresponding Authors Dieter Egli 1150 St. Nicholas Avenue New York, NY 10032 [email protected] Lori Sussel 1775 Aurora Ct. Aurora, CO 80045 [email protected] Word Count: Abstract= 149; Body= 4773 Total Paper Figures= 7, Total Supplemental Tables= 4, Total Supplemental Figures= 5 Diabetes Publish Ahead of Print, published online September 13, 2019 Diabetes Page 2 of 105 Abstract Inactivation of the β cell transcription factor NEUROD1 causes diabetes in mice and humans. In this study, we uncovered novel functions of Neurod1 during murine islet cell development and during the differentiation of human embryonic stem cells (HESCs) into insulin-producing cells. In mice, we determined that Neurod1 is required for perinatal proliferation of alpha and beta cells. Surprisingly, apoptosis only makes a minor contribution to beta cell loss when Neurod1 is deleted. Inactivation of NEUROD1 in HESCs severely impaired their differentiation from pancreatic progenitors into insulin expressing (HESC-beta) cells; however survival or proliferation was not affected at the time points analyzed. NEUROD1 was also required in HESC-beta cells for the full activation of an essential beta cell transcription factor network. -

Immunohorizon 2100067 Suppl
Supplementary Table 1: Differentially expressed genes in L group versus vehicle-treated group (1004 genes) Upregulated genes Downregulated genes Il33 4930438A08Rik Etv4 Mcoln2 Gpr18 Cyth3 Mad2l1 Ttc38 Rab19 Gpd1 Klhl6 Ccl5 Nlrp3 Clec4e Ddx58 Bcl2a1d Sh3pxd2b Map3k6 Rab39 Calr3 Ptger4 Fndc10 Serpinb2 Pfkfb3 Dgkh Spata13 Ptger2 Itgav Ckb Hist1h1c Gm12522 Susd3 Pard6g Lif Ccl2 Dusp1 Vgf Gm14286 Spred1 Brca2 Ncapd2 Tle2 Sorbs1 4933431G14Rik Il6 Oasl2 Smim3 Gm12059 Rnf186 Maml2 Gm16576 Brip1 Lfng Smc1b Morn5 Akap12 Gdnf Furin Vegfa Fgr Samsn1 Bag2 Mtfp1 Aurkb Sorbs3 Fam13c Edn1 Styk1 Csrnp1 6030443J06Rik Samd9l Nod2 Tmem218 Plk1 Neurl1b Tmem79 Sarm1 U90926 Gm26675 Sema3e Ifi202b Il10ra Clic4 Tsc22d3 Ttc28 Oip5 Dixdc1 Rarb Il1b Gbp2 Dtx2 Gm7609 Trim25 Gm17275 Fut10 Bmf Knstrn Zfp108 Gm13710 Tpbg Peli1 Gm11496 Adamts7 Slfn4 Ksr1 Zfp760 H2-T10 6430548M08Rik Olfml2b AC159314.1 Cxcl10 Zfp36 Nfkbie Gm9774 Irf9 Gstt1 Kcnn1 Rnasel Pax6 Tbc1d8 9930111J21Rik2 Csf2 Trim30b Tmem171 Lta Ccr1 Hck Chst12 Nfia Cerkl Kcnab3 Fam171a2 Sox30 Lix1 Gm20219 Gja1 Pmp22 Tnfsf13b Tmem43 Casp3 Lrrc56 Coq8a Mtl5 Sprr2d Hmx3 Klk9 Ptch1 Zc3h12a Pdlim7 Gm16675 Foxm1 Cdon Myo7b Gdf9 Rsad2 Cxcl11 Kdm6b Junb Procr Hcls1 Pdcd4 Agmo Catsperg1 Fbxo5 Speer9-ps1 Rtp4 Col5a3 Sema4c Eid3 Micall2 6230400D17Rik Ak1 Col4a6 4930558J18Rik Daglb BC037032 Ttc39c Fam46c Oaf Tnip1 Fam214b Tgfb1 Il6ra Rab3d Nav2 Rnf144b Mkln1os Ifit1 Fam110c Glrx Sod2 Pilra Dusp10 Hist2h2be Lpar5 Arrb1 Fam46b AC153365.1 Isg15 Hist2h2aa2 Dusp2 Arhgef3 Itga5 Casp6 Brca1 Racgap1 Depdc1b Ppp1r1a Snhg7os Oasl1 -

Comparative Analysis of Two C-Terminal Kinesin Motor Proteins: KIFC1 and KIFC5A
Cell Motility and the Cytoskeleton 58:213–230 (2004) Comparative Analysis of Two C-Terminal Kinesin Motor Proteins: KIFC1 and KIFC5A Yuguo Zhang and Ann O. Sperry* Department of Anatomy and Cell Biology, Brody School of Medicine at East Carolina University, Greenville, North Carolina We have taken advantage of the close structural relationship between two C- terminal motors, KIFC5A and KIFC1, to examine the sequence requirements for targeting of these two motors within the cell. Although KIFC5A and KIFC1 are almost identical in their motor and stalk domains, they differ in well-defined regions of their tail domains. Specific antisera to these motors were used to determine their localization to distinct subcellular compartments, the spindle for KIFC5A or membranous organelles for KIFC1. In addition to defining the intracellular localization of KIFC1, the reactivity of the KIFC1 antibody demon- strates that this motor contains a frame shift with respect to KIFC5A and is likely the product of a separate gene. The divergent tail domains of these motors are predicted to harbor specific information that directs them to their correct intra- cellular targets. In order to define the sequences responsible for the differential localization of these two motors, GFP was fused to motors with various tail deletions and their localization visualized after transfection. We were able to identify distinct sequences in each motor responsible for its unique cellular localization. The KIFC5A tail contains a 43 amino acid sequence with both nuclear localization and microtubule binding activity while KIFC1 contains a 19 amino acid sequence sufficient to target this motor to membrane-bounded or- ganelles.