Phosphorylation of Titin Affecting the Mechanical Function of Cardiomyocytes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Identification of Potential Markers for Type 2 Diabetes Mellitus Via Bioinformatics Analysis
1868 MOLECULAR MEDICINE REPORTS 22: 1868-1882, 2020 Identification of potential markers for type 2 diabetes mellitus via bioinformatics analysis YANA LU1, YIHANG LI1, GUANG LI1* and HAITAO LU2* 1Key Laboratory of Dai and Southern Medicine of Xishuangbanna Dai Autonomous Prefecture, Yunnan Branch, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Jinghong, Yunnan 666100; 2Key Laboratory of Systems Biomedicine, Shanghai Center for Systems Biomedicine, Shanghai Jiao Tong University, Shanghai 200240, P.R. China Received March 20, 2019; Accepted January 20, 2020 DOI: 10.3892/mmr.2020.11281 Abstract. Type 2 diabetes mellitus (T2DM) is a multifactorial and cell proliferation’. These candidate genes were also involved in multigenetic disease, and its pathogenesis is complex and largely different signaling pathways associated with ‘PI3K/Akt signaling unknown. In the present study, microarray data (GSE201966) of pathway’, ‘Rap1 signaling pathway’, ‘Ras signaling pathway’ β-cell enriched tissue obtained by laser capture microdissection and ‘MAPK signaling pathway’, which are highly associated were downloaded, including 10 control and 10 type 2 diabetic with the development of T2DM. Furthermore, a microRNA subjects. A comprehensive bioinformatics analysis of microarray (miR)-target gene regulatory network and a transcription data in the context of protein-protein interaction (PPI) networks factor-target gene regulatory network were constructed based was employed, combined with subcellular location information on miRNet and NetworkAnalyst databases, respectively. to mine the potential candidate genes for T2DM and provide Notably, hsa-miR‑192-5p, hsa-miR‑124-5p and hsa-miR‑335-5p further insight on the possible mechanisms involved. First, appeared to be involved in T2DM by potentially regulating the differential analysis screened 108 differentially expressed expression of various candidate genes, including procollagen genes. -

Genome-Wide Linkage and Admixture Mapping of Type 2 Diabetes In
ORIGINAL ARTICLE Genome-Wide Linkage and Admixture Mapping of Type 2 Diabetes in African American Families From the American Diabetes Association GENNID (Genetics of NIDDM) Study Cohort Steven C. Elbein,1,2 Swapan K. Das,1,2 D. Michael Hallman,3 Craig L. Hanis,3 and Sandra J. Hasstedt4 OBJECTIVE—We used a single nucleotide polymorphism (SNP) map in a large cohort of 580 African American families to identify regions linked to type 2 diabetes, age of type 2 diabetes ype 2 diabetes is marked by a clear genetic diagnosis, and BMI. propensity, a high concordance in identical twins, tendencies for both diabetes and age of RESEARCH DESIGN AND METHODS—After removing outli- onset to be familial (1), and marked differences ers and problematic samples, we conducted linkage analysis T in prevalence among ethnic groups (2). Despite consider- using 5,914 SNPs in 1,344 individuals from 530 families. Linkage analysis was conducted using variance components for type 2 able evidence for a genetic predisposition, unraveling the diabetes, age of type 2 diabetes diagnosis, and BMI and nonpara- genetic etiology has been daunting, with few confirmed metric linkage analyses. Ordered subset analyses were con- genes identified from genome-wide linkage scans. Recent ducted ranking on age of type 2 diabetes diagnosis, BMI, waist successes with genome-wide association scans (3) have circumference, waist-to-hip ratio, and amount of European ad- greatly increased the number of confirmed genetic loci, mixture. Admixture mapping was conducted using 4,486 markers but these successes have been limited primarily to Cauca- not in linkage disequilibrium. -

The Adaptation and Changes of Titin System Following Exercise Training Hassan Farhadi 1 and Mir Hojjat Mosavineghad 2
Available online a t www.pelagiaresearchlibrary.com Pelagia Research Library European Journal of Experimental Biology, 2012, 2 (4):1256-1260 ISSN: 2248 –9215 CODEN (USA): EJEBAU The adaptation and changes of titin system following exercise training Hassan Farhadi 1 and Mir Hojjat Mosavineghad 2 1Department of Physical Education and Sport Sciences, Ahar branch, Islamic Azad University, Ahar, Iran 2Department of Physical Education and Sport Sciences, khoy branch, Islamic Azad University, khoy, Iran _____________________________________________________________________________________________ ABSTRACT Titin is a large structural protein in muscle that has the ability to store elastic energy. The specific structure of titin and its elastic nature has recently become better understood. In addition, the utilization of stored elastic energy in human movement and the significant contribution of not only tendon but muscle tissue itself to this process has been re-evaluated. In this study, we reviewed the effects of exercise training on titin system (titin expression and titin- complex proteins). Keywords: Titin, titin-complex proteins, exercise training _________________________________________________________________________________________ The giant sarcomeric protein titin is 3-4 MDa and spans the sarcomere from Z-line to M-line. Titin was first discovered in 1979 by Wang et. al. (1), who initially thought that titin was a collection of polypeptides that formed one large protein. Although titin’s physical identification eluded researchers for many years, probably due to its susceptibility to proteolytic cleavage, many scientists, including Earnest Starling and A.F. Huxley, posited its existence (2). Starling and Huxley modeled their theories on the premise that something within striated muscle was regulating passive properties. This is now known to be the role of titin, which is one peptide, encoded by a single gene, TTN (2). -

A Population-Specific Major Allele Reference Genome from the United
Edith Cowan University Research Online ECU Publications Post 2013 2021 A population-specific major allele efr erence genome from the United Arab Emirates population Gihan Daw Elbait Andreas Henschel Guan K. Tay Edith Cowan University Habiba S. Al Safar Follow this and additional works at: https://ro.ecu.edu.au/ecuworkspost2013 Part of the Life Sciences Commons, and the Medicine and Health Sciences Commons 10.3389/fgene.2021.660428 Elbait, G. D., Henschel, A., Tay, G. K., & Al Safar, H. S. (2021). A population-specific major allele reference genome from the United Arab Emirates population. Frontiers in Genetics, 12, article 660428. https://doi.org/10.3389/ fgene.2021.660428 This Journal Article is posted at Research Online. https://ro.ecu.edu.au/ecuworkspost2013/10373 fgene-12-660428 April 19, 2021 Time: 16:18 # 1 ORIGINAL RESEARCH published: 23 April 2021 doi: 10.3389/fgene.2021.660428 A Population-Specific Major Allele Reference Genome From The United Arab Emirates Population Gihan Daw Elbait1†, Andreas Henschel1,2†, Guan K. Tay1,3,4,5 and Habiba S. Al Safar1,3,6* 1 Center for Biotechnology, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates, 2 Department of Electrical Engineering and Computer Science, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates, 3 Department of Biomedical Engineering, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates, 4 Division of Psychiatry, Faculty of Health and Medical Sciences, The University of Western Australia, Crawley, WA, Australia, 5 School of Medical and Health Sciences, Edith Cowan University, Joondalup, WA, Australia, 6 Department of Genetics and Molecular Biology, College of Medicine and Health Sciences, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates The ethnic composition of the population of a country contributes to the uniqueness of each national DNA sequencing project and, ideally, individual reference genomes are required to reduce the confounding nature of ethnic bias. -

Regulation of Titin-Based Cardiac Stiffness by Unfolded Domain Oxidation (Undox)
Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx) Christine M. Loeschera,1, Martin Breitkreuzb,1, Yong Lia, Alexander Nickelc, Andreas Ungera, Alexander Dietld, Andreas Schmidte, Belal A. Mohamedf, Sebastian Kötterg, Joachim P. Schmitth, Marcus Krügere,i, Martina Krügerg, Karl Toischerf, Christoph Maackc, Lars I. Leichertj, Nazha Hamdanib, and Wolfgang A. Linkea,2 aInstitute of Physiology II, University of Munster, 48149 Munster, Germany; bInstitute of Physiology, Ruhr University Bochum, 44801 Bochum, Germany; cComprehensive Heart Failure Center Wuerzburg, University Clinic Wuerzburg, 97078 Wuerzburg, Germany; dDepartment of Internal Medicine II, University Hospital Regensburg, 93053 Regensburg, Germany; eInstitute for Genetics, University of Cologne, 50931 Cologne, Germany; fDepartment of Cardiology and Pneumology, University Medicine Goettingen, 37075 Goettingen, Germany; gDepartment of Cardiovascular Physiology, Heinrich Heine University, 40225 Düsseldorf, Germany; hDepartment of Pharmacology and Clinical Pharmacology, Heinrich Heine University, 40225 Düsseldorf, Germany; iCenter for Molecular Medicine and Excellence Cluster "Cellular Stress Responses in Aging-Associated Diseases" (CECAD), University of Cologne, 50931 Cologne, Germany; and jInstitute for Biochemistry and Pathobiochemistry, Ruhr University Bochum, 44801 Bochum, Germany Edited by Jonathan Seidman, Harvard University, Boston, MA, and approved August 12, 2020 (received for review March 14, 2020) The relationship between oxidative stress and -

Postmortem Changes in the Myofibrillar and Other Cytoskeletal Proteins in Muscle
BIOCHEMISTRY - IMPACT ON MEAT TENDERNESS Postmortem Changes in the Myofibrillar and Other C'oskeletal Proteins in Muscle RICHARD M. ROBSON*, ELISABETH HUFF-LONERGAN', FREDERICK C. PARRISH, JR., CHIUNG-YING HO, MARVIN H. STROMER, TED W. HUIATT, ROBERT M. BELLIN and SUZANNE W. SERNETT introduction filaments (titin), and integral Z-line region (a-actinin, Cap Z), as well as proteins of the intermediate filaments (desmin, The cytoskeleton of "typical" vertebrate cells contains paranemin, and synemin), Z-line periphery (filamin) and three protein filament systems, namely the -7-nm diameter costameres underlying the cell membrane (filamin, actin-containing microfilaments, the -1 0-nm diameter in- dystrophin, talin, and vinculin) are listed along with an esti- termediate filaments (IFs), and the -23-nm diameter tubu- mate of their abundance, approximate molecular weights, lin-containing microtubules (Robson, 1989, 1995; Robson and number of subunits per molecule. Because the myofibrils et al., 1991 ).The contractile myofibrils, which are by far the are the overwhelming components of the skeletal muscle cell major components of developed skeletal muscle cells and cytoskeleton, the approximate percentages of the cytoskel- are responsible for most of the desirable qualities of muscle eton listed for the myofibrillar proteins (e.g., myosin, actin, foods (Robson et al., 1981,1984, 1991 1, can be considered tropomyosin, a-actinin, etc.) also would represent their ap- the highly expanded corollary of the microfilament system proximate percentages of total myofibrillar protein. of non-muscle cells. The myofibrils, IFs, cell membrane skel- eton (complex protein-lattice subjacent to the sarcolemma), Some Important Characteristics, Possible and attachment sites connecting these elements will be con- Roles, and Postmortem Changes of Key sidered as comprising the muscle cell cytoskeleton in this Cytoskeletal Proteins review. -

Α-Actinin/Titin Interaction: a Dynamic and Mechanically Stable Cluster of Bonds in the Muscle Z-Disk
α-Actinin/titin interaction: A dynamic and mechanically stable cluster of bonds in the muscle Z-disk Marco Grisona, Ulrich Merkela, Julius Kostanb, Kristina Djinovic-Carugob,c, and Matthias Riefa,d,1 aPhysik Department E22, Technische Universität München, 85748 Garching, Germany; bDepartment of Structural and Computational Biology, Max F. Perutz Laboratories, University of Vienna, A-1030 Vienna, Austria; cDepartment of Biochemistry, Faculty of Chemistry and Chemical Technology, University of Ljubljana, SI-1000 Ljubljana, Slovenia; and dMunich Center for Integrated Protein Science, 81377 Munich, Germany Edited by James A. Spudich, Stanford University School of Medicine, Stanford, CA, and approved December 16, 2016 (received for review August 2, 2016) Stable anchoring of titin within the muscle Z-disk is essential for In humans, the isoforms of titin exhibit four to seven Z-repeats preserving muscle integrity during passive stretching. One of the (15, 16, 20). The structure of the EF3-4 hands complex with titin main candidates for anchoring titin in the Z-disk is the actin cross- Z-repeat 7 shows the bound Z-repeat in an α-helical confor- linker α-actinin. The calmodulin-like domain of α-actinin binds to mation (21). In solution assays, binding affinities of various the Z-repeats of titin. However, the mechanical and kinetic prop- Z-repeats to EF3-4 were determined to lie in the micromolar erties of this important interaction are still unknown. Here, we use range (22). Micromolar affinity points only to a moderately a dual-beam optical tweezers assay to study the mechanics of this stable interaction, the kinetics of which are unknown. -
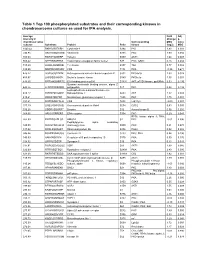
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Association of Titin and Myosin Heavy Chain in Developing Skeletal Muscle (Myogenesis/Cytoskeleton/Assembly in Vvo) W
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 74%-7500, August 1992 Cell Biology Association of titin and myosin heavy chain in developing skeletal muscle (myogenesis/cytoskeleton/assembly in vvo) W. B. ISAACS*, I. S. KIM, A. STRUVE, AND A. B. FULTONt Department of Biochemistry, University of Iowa, Iowa City, IA 52242 Communicated by Sheldon Penman, May 22, 1992 ABSTRACT To understand molecular interactions that deficient medium (10). After labeling, cultures either were organize developing myoflbrils, we examined the biosynthesis extracted immediately or were chased by adding complete and interaction of titin and myosin heavy chain in cultures of medium supplemented with 2 mM unlabeled methionine and developing muscle. Use of pulse-labeling, immunoprecipita- incubating at 370C for various times before extraction. Ex- tion, and a reversible cross-linking procedure demonstrates tractions used 0.5% Triton X-100 in extraction buffer (100 that within minutes of synthesis, titin and myosin heavy chain mM KCI/10 mM Pipes, pH 6.8/300 mM sucrose/2 mM can be chemically cross-linked into very large, detergent- MgCI2/1 mM EGTA) containing protease inhibitors (1 mM resistant complexes retaining many features of intact myo- phenylmethylsulfonyl fluoride and 100 mM leupeptin; ref. tubes. These complexes, predominantly of titin and myosin, 11). occur very early in myofibrillogenesis as well as later. These Immunoprecipitation. Titin was precipitated by using a data suggest that synthesis and assembly oftitin and myosin are mouse monoclonal antibody (mAb), AMF-1, as described temporally and spatially coordinated in nascent myofibrils and (10). Muscle-specific myosin heavy chain (hereafter myosin) support the hypothesis that titin molecules help to organize was precipitated with mAb MF-20 (12), a gift ofD. -

Titin N2A Domain and Its Interactions at the Sarcomere
International Journal of Molecular Sciences Review Titin N2A Domain and Its Interactions at the Sarcomere Adeleye O. Adewale and Young-Hoon Ahn * Department of Chemistry, Wayne State University, Detroit, MI 48202, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-(313)-577-1384 Abstract: Titin is a giant protein in the sarcomere that plays an essential role in muscle contraction with actin and myosin filaments. However, its utility goes beyond mechanical functions, extending to versatile and complex roles in sarcomere organization and maintenance, passive force, mechanosens- ing, and signaling. Titin’s multiple functions are in part attributed to its large size and modular structures that interact with a myriad of protein partners. Among titin’s domains, the N2A element is one of titin’s unique segments that contributes to titin’s functions in compliance, contraction, structural stability, and signaling via protein–protein interactions with actin filament, chaperones, stress-sensing proteins, and proteases. Considering the significance of N2A, this review highlights structural conformations of N2A, its predisposition for protein–protein interactions, and its multiple interacting protein partners that allow the modulation of titin’s biological effects. Lastly, the nature of N2A for interactions with chaperones and proteases is included, presenting it as an important node that impacts titin’s structural and functional integrity. Keywords: titin; N2A domain; protein–protein interaction 1. Introduction Citation: Adewale, A.O.; Ahn, Y.-H. The complexity of striated muscle is defined by the intricate organization of its com- Titin N2A Domain and Its ponents [1]. The involuntary cardiac and voluntary skeletal muscles are the primary types Interactions at the Sarcomere. -

B3GALNT2 Is a Gene Associated with Congenital Muscular Dystrophy with Brain Malformations
European Journal of Human Genetics (2014) 22, 707–710 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg SHORT REPORT B3GALNT2 is a gene associated with congenital muscular dystrophy with brain malformations Carola Hedberg*,1, Anders Oldfors1 and Niklas Darin2 Congenital muscular dystrophies associated with brain malformations are a group of disorders frequently associated with aberrant glycosylation of a-dystroglycan. They include disease entities such a Walker–Warburg syndrome, muscle–eye–brain disease and various other clinical phenotypes. Different genes involved in glycosylation of a-dystroglycan are associated with these dystroglycanopathies. We describe a 5-year-old girl with psychomotor retardation, ataxia, spasticity, muscle weakness and increased serum creatine kinase levels. Immunhistochemistry of skeletal muscle revealed reduced glycosylated a-dystroglycan. Magnetic resonance imaging of the brain at 3.5 years of age showed increased T2 signal from supratentorial and infratentorial white matter, a hypoplastic pons and subcortical cerebellar cysts. By whole exome sequencing, the patient was identified to be compound heterozygous for a one-base duplication and a missense mutation in the gene B3GALNT2 (b-1,3-N-acetylgalactos- aminyltransferase 2; B3GalNAc-T2). This patient showed a milder phenotype than previously described patients with mutations in the B3GALNT2 gene. European Journal of Human Genetics (2014) 22, 707–710; doi:10.1038/ejhg.2013.223; published online 2 October 2013 Keywords: