Expression Profile and Bioinformatics Analysis of Circular Rnas in Acute
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

The Basal Bodies of Chlamydomonas Reinhardtii Susan K
Dutcher and O’Toole Cilia (2016) 5:18 DOI 10.1186/s13630-016-0039-z Cilia REVIEW Open Access The basal bodies of Chlamydomonas reinhardtii Susan K. Dutcher1* and Eileen T. O’Toole2 Abstract The unicellular green alga, Chlamydomonas reinhardtii, is a biflagellated cell that can swim or glide. C. reinhardtii cells are amenable to genetic, biochemical, proteomic, and microscopic analysis of its basal bodies. The basal bodies contain triplet microtubules and a well-ordered transition zone. Both the mother and daughter basal bodies assemble flagella. Many of the proteins found in other basal body-containing organisms are present in the Chlamydomonas genome, and mutants in these genes affect the assembly of basal bodies. Electron microscopic analysis shows that basal body duplication is site-specific and this may be important for the proper duplication and spatial organization of these organelles. Chlamydomonas is an excellent model for the study of basal bodies as well as the transition zone. Keywords: Site-specific basal body duplication, Cartwheel, Transition zone, Centrin fibers Phylogeny and conservation of proteins Centrin, SPD2/CEP192, Asterless/CEP152; CEP70, The green lineage or Viridiplantae consists of the green delta-tubulin, and epsilon-tubulin. Chlamydomonas has algae, which include Chlamydomonas, the angiosperms homologs of all of these based on sequence conservation (the land plants), and the gymnosperms (conifers, cycads, except PLK4, CEP152, and CEP192. Several lines of evi- ginkgos). They are grouped together because they have dence suggests that CEP152, CEP192, and PLK4 interact chlorophyll a and b and lack phycobiliproteins. The green [20, 52] and their concomitant absence in several organ- algae together with the cycads and ginkgos have basal isms suggests that other mechanisms exist that allow for bodies and cilia, while the angiosperms and conifers have control of duplication [4]. -

The Endocytic Membrane Trafficking Pathway Plays a Major Role
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of Liverpool Repository RESEARCH ARTICLE The Endocytic Membrane Trafficking Pathway Plays a Major Role in the Risk of Parkinson’s Disease Sara Bandres-Ciga, PhD,1,2 Sara Saez-Atienzar, PhD,3 Luis Bonet-Ponce, PhD,4 Kimberley Billingsley, MSc,1,5,6 Dan Vitale, MSc,7 Cornelis Blauwendraat, PhD,1 Jesse Raphael Gibbs, PhD,7 Lasse Pihlstrøm, MD, PhD,8 Ziv Gan-Or, MD, PhD,9,10 The International Parkinson’s Disease Genomics Consortium (IPDGC), Mark R. Cookson, PhD,4 Mike A. Nalls, PhD,1,11 and Andrew B. Singleton, PhD1* 1Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, USA 2Instituto de Investigación Biosanitaria de Granada (ibs.GRANADA), Granada, Spain 3Transgenics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, USA 4Cell Biology and Gene Expression Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, USA 5Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, University of Liverpool, Liverpool, United Kingdom 6Department of Pathophysiology, University of Tartu, Tartu, Estonia 7Computational Biology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, USA 8Department of Neurology, Oslo University Hospital, Oslo, Norway 9Department of Neurology and Neurosurgery, Department of Human Genetics, McGill University, Montréal, Quebec, Canada 10Department of Neurology and Neurosurgery, Montreal Neurological Institute, McGill University, Montréal, Quebec, Canada 11Data Tecnica International, Glen Echo, Maryland, USA ABSTRACT studies, summary-data based Mendelian randomization Background: PD is a complex polygenic disorder. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Identification of Transcriptomic Differences Between Lower
International Journal of Molecular Sciences Article Identification of Transcriptomic Differences between Lower Extremities Arterial Disease, Abdominal Aortic Aneurysm and Chronic Venous Disease in Peripheral Blood Mononuclear Cells Specimens Daniel P. Zalewski 1,*,† , Karol P. Ruszel 2,†, Andrzej St˛epniewski 3, Dariusz Gałkowski 4, Jacek Bogucki 5 , Przemysław Kołodziej 6 , Jolanta Szyma ´nska 7 , Bartosz J. Płachno 8 , Tomasz Zubilewicz 9 , Marcin Feldo 9,‡ , Janusz Kocki 2,‡ and Anna Bogucka-Kocka 1,‡ 1 Chair and Department of Biology and Genetics, Medical University of Lublin, 4a Chod´zkiSt., 20-093 Lublin, Poland; [email protected] 2 Chair of Medical Genetics, Department of Clinical Genetics, Medical University of Lublin, 11 Radziwiłłowska St., 20-080 Lublin, Poland; [email protected] (K.P.R.); [email protected] (J.K.) 3 Ecotech Complex Analytical and Programme Centre for Advanced Environmentally Friendly Technologies, University of Marie Curie-Skłodowska, 39 Gł˛ebokaSt., 20-612 Lublin, Poland; [email protected] 4 Department of Pathology and Laboratory Medicine, Rutgers-Robert Wood Johnson Medical School, One Robert Wood Johnson Place, New Brunswick, NJ 08903-0019, USA; [email protected] 5 Chair and Department of Organic Chemistry, Medical University of Lublin, 4a Chod´zkiSt., Citation: Zalewski, D.P.; Ruszel, K.P.; 20-093 Lublin, Poland; [email protected] St˛epniewski,A.; Gałkowski, D.; 6 Laboratory of Diagnostic Parasitology, Chair and Department of Biology and Genetics, Medical University of Bogucki, J.; Kołodziej, P.; Szyma´nska, Lublin, 4a Chod´zkiSt., 20-093 Lublin, Poland; [email protected] J.; Płachno, B.J.; Zubilewicz, T.; Feldo, 7 Department of Integrated Paediatric Dentistry, Chair of Integrated Dentistry, Medical University of Lublin, M.; et al. -

Is Cep70, a Centrosomal Protein with New Roles in Breast Cancer Dissemination and Metastasis, a Facilitator of Epithelial-Mesenchymal Transition?
Is Cep70, a centrosomal protein with new roles in breast cancer dissemination and metastasis, a facilitator of epithelial-mesenchymal transition? Pedro A. Lazo 1,2 1 Experimental Therapeutics and Translational Oncology Program, Instituto de Biología Molecular y Celular del Cáncer, Consejo Superior de Investigaciones Científicas (CSIC), Universidad de Salamanca, Salamanca, Spain 2 Instituto de Investigación Biomédica de Salamanca (IBSAL), Hospital Universitario de Salamanca, Salamanca, Spain Running title: Cep70 and EMT Disclosures: None declared. Contact address: [email protected] 1 Introduction Microtubules are driving mechanisms of chromosomes, intracellular organelle movement, and cell shape and motility. Microtubules are organized on centrosomes, which are assembled in microtubule-organizing centers (MTOC). In mitosis there is no nuclear envelope and centromeres associated to microtubules are mainly involved in chromosome redistribution into daughter cells. In differentiated cells the microtubule- organizing centers are dispersed in the cytoplasm (non centrosomal (ncMTOC)) and interact with the minus end of microtubules through γ-tubulin. 1 However, it is not known if the microtubule contribution to tumor biology is only by facilitating tumor aneuploidy. During tumor dissemination, a process not linked to cell division, important changes take place in cell shape and motility. Microtubules are very dynamic because of their inherent structural instability, and this plasticity facilitates their reorganization during the epithelial-mesenchymal -

Autism-Associated Mir-873 Regulates ARID1B, SHANK3 and NRXN2
Lu et al. Translational Psychiatry (2020) 10:418 https://doi.org/10.1038/s41398-020-01106-8 Translational Psychiatry ARTICLE Open Access Autism-associated miR-873 regulates ARID1B, SHANK3 and NRXN2 involved in neurodevelopment Jing Lu 1, Yan Zhu 1, Sarah Williams2, Michelle Watts 3,MaryA.Tonta1, Harold A. Coleman1, Helena C. Parkington1 and Charles Claudianos 1,4 Abstract Autism spectrum disorders (ASD) are highly heritable neurodevelopmental disorders with significant genetic heterogeneity. Noncoding microRNAs (miRNAs) are recognised as playing key roles in development of ASD albeit the function of these regulatory genes remains unclear. We previously conducted whole-exome sequencing of Australian families with ASD and identified four novel single nucleotide variations in mature miRNA sequences. A pull-down transcriptome analysis using transfected SH-SY5Y cells proposed a mechanistic model to examine changes in binding affinity associated with a unique mutation found in the conserved ‘seed’ region of miR-873-5p (rs777143952: T > A). Results suggested several ASD-risk genes were differentially targeted by wild-type and mutant miR-873 variants. In the current study, a dual-luciferase reporter assay confirmed miR-873 variants have a 20-30% inhibition/dysregulation effect on candidate autism risk genes ARID1B, SHANK3 and NRXN2 and also confirmed the affected expression with qPCR. In vitro mouse hippocampal neurons transfected with mutant miR-873 showed less morphological complexity and enhanced sodium currents and excitatory neurotransmission compared to cells transfected with wild-type miR- 873. A second in vitro study showed CRISPR/Cas9 miR-873 disrupted SH-SY5Y neuroblastoma cells acquired a neuronal-like morphology and increased expression of ASD important genes ARID1B, SHANK3, ADNP2, ANK2 and CHD8. -

FIST/HIPK3: a Fas/FADD-Interacting Serine/Threonine Kinase That Induces FADD Phosphorylation and Inhibits Fas-Mediated Jun NH2-Terminal Kinase Activation
FIST/HIPK3: a Fas/FADD-interacting Serine/Threonine Kinase that Induces FADD Phosphorylation and Inhibits Fas-mediated Jun NH2-terminal Kinase Activation By Véronique Rochat-Steiner, Karin Becker, Olivier Micheau, Pascal Schneider, Kim Burns, and Jürg Tschopp From the Institute of Biochemistry, University of Lausanne, BIL Biomedical Research Center, CH- 1066 Epalinges, Switzerland Abstract Fas is a cell surface death receptor that signals apoptosis. Several proteins have been identified that bind to the cytoplasmic death domain of Fas. Fas-associated death domain (FADD), which couples Fas to procaspase-8, and Daxx, which couples Fas to the Jun NH2-terminal kinase pathway, bind independently to the Fas death domain. We have identified a 130-kD ki- nase designated Fas-interacting serine/threonine kinase/homeodomain-interacting protein ki- nase (FIST/HIPK3) as a novel Fas-interacting protein. Binding to Fas is mediated by a con- served sequence in the COOH terminus of the protein. FIST/HIPK3 is widely expressed in mammalian tissues and is localized both in the nucleus and in the cytoplasm. In transfected cell lines, FIST/HIPK3 causes FADD phosphorylation, thereby promoting FIST/HIPK3–FADD– Fas interaction. Although Fas ligand–induced activation of Jun NH2-terminal kinase is im- paired by overexpressed active FIST/HIPK3, cell death is not affected. These results suggest that Fas-associated FIST/HIPK3 modulates one of the two major signaling pathways of Fas. Key words: Fas/CD95 • apoptosis • kinase • Jun NH2-terminal kinase • signal transduction Introduction Fas (APO-1/CD95) is a member of the TNFR family and tiate apoptosis by subsequent cleavage of downstream ef- induces apoptosis when cross-linked with Fas ligand (FasL)1 fector caspases (caspase-3, -6, and -7). -

Mechanisms Underlying Phenotypic Heterogeneity in Simplex Autism Spectrum Disorders
Mechanisms Underlying Phenotypic Heterogeneity in Simplex Autism Spectrum Disorders Andrew H. Chiang Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2021 © 2021 Andrew H. Chiang All Rights Reserved Abstract Mechanisms Underlying Phenotypic Heterogeneity in Simplex Autism Spectrum Disorders Andrew H. Chiang Autism spectrum disorders (ASD) are a group of related neurodevelopmental diseases displaying significant genetic and phenotypic heterogeneity. Despite recent progress in ASD genetics, the nature of phenotypic heterogeneity across probands is not well understood. Notably, likely gene- disrupting (LGD) de novo mutations affecting the same gene often result in substantially different ASD phenotypes. We find that truncating mutations in a gene can result in a range of relatively mild decreases (15-30%) in gene expression due to nonsense-mediated decay (NMD), and show that more severe autism phenotypes are associated with greater decreases in expression. We also find that each gene with recurrent ASD mutations can be described by a parameter, phenotype dosage sensitivity (PDS), which characteriZes the relationship between changes in a gene’s dosage and changes in a given phenotype. Using simple linear models, we show that changes in gene dosage account for a substantial fraction of phenotypic variability in ASD. We further observe that LGD mutations affecting the same exon frequently lead to strikingly similar phenotypes in unrelated ASD probands. These patterns are observed for two independent proband cohorts and multiple important ASD-associated phenotypes. The observed phenotypic similarities are likely mediated by similar changes in gene dosage and similar perturbations to the relative expression of splicing isoforms. -

Transcript Variants of Genes Involved in Neurodegeneration Are Differentially Regulated by the APOE and MAPT Haplotypes
G C A T T A C G G C A T genes Article Transcript Variants of Genes Involved in Neurodegeneration Are Differentially Regulated by the APOE and MAPT Haplotypes Sulev Koks 1,2,* , Abigail L. Pfaff 1,2, Vivien J. Bubb 3 and John P. Quinn 3 1 Perron Institute for Neurological and Translational Science, Perth, WA 6009, Australia; [email protected] 2 Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Perth, WA 6150, Australia 3 Department of Pharmacology and Therapeutics, Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool L69 3BX, UK; [email protected] (V.J.B.); [email protected] (J.P.Q.) * Correspondence: [email protected]; Tel.: +61-(0)-8-6457-0313 Abstract: Genetic variations at the Apolipoprotein E (ApoE) and microtubule-associated protein tau (MAPT) loci have been implicated in multiple neurogenerative diseases, but their exact molecular mechanisms are unclear. In this study, we performed transcript level linear modelling using the blood whole transcriptome data and genotypes of the 570 subjects in the Parkinson’s Progression Markers Initiative (PPMI) cohort. ApoE, MAPT haplotypes and two SNPs at the SNCA locus (rs356181, rs3910105) were used to detect expression quantitative trait loci eQTLs associated with the transcriptome and differential usage of transcript isoforms. As a result, we identified 151 genes associated with the genotypic variations, 29 cis and 122 trans eQTL positions. Profound effect with genome-wide significance of ApoE e4 haplotype on the expression of TOMM40 transcripts was identified. This finding potentially explains in part the frequently established genetic association Citation: Koks, S.; Pfaff, A.L.; Bubb, with the APOE e4 haplotypes in neurodegenerative diseases. -
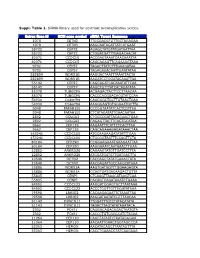
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo