Examination of the Expression and Prognostic Significance of Dlgaps in Gastric Cancer Using the TCGA Database and Bioinformatic Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Autism-Associated Mir-873 Regulates ARID1B, SHANK3 and NRXN2
Lu et al. Translational Psychiatry (2020) 10:418 https://doi.org/10.1038/s41398-020-01106-8 Translational Psychiatry ARTICLE Open Access Autism-associated miR-873 regulates ARID1B, SHANK3 and NRXN2 involved in neurodevelopment Jing Lu 1, Yan Zhu 1, Sarah Williams2, Michelle Watts 3,MaryA.Tonta1, Harold A. Coleman1, Helena C. Parkington1 and Charles Claudianos 1,4 Abstract Autism spectrum disorders (ASD) are highly heritable neurodevelopmental disorders with significant genetic heterogeneity. Noncoding microRNAs (miRNAs) are recognised as playing key roles in development of ASD albeit the function of these regulatory genes remains unclear. We previously conducted whole-exome sequencing of Australian families with ASD and identified four novel single nucleotide variations in mature miRNA sequences. A pull-down transcriptome analysis using transfected SH-SY5Y cells proposed a mechanistic model to examine changes in binding affinity associated with a unique mutation found in the conserved ‘seed’ region of miR-873-5p (rs777143952: T > A). Results suggested several ASD-risk genes were differentially targeted by wild-type and mutant miR-873 variants. In the current study, a dual-luciferase reporter assay confirmed miR-873 variants have a 20-30% inhibition/dysregulation effect on candidate autism risk genes ARID1B, SHANK3 and NRXN2 and also confirmed the affected expression with qPCR. In vitro mouse hippocampal neurons transfected with mutant miR-873 showed less morphological complexity and enhanced sodium currents and excitatory neurotransmission compared to cells transfected with wild-type miR- 873. A second in vitro study showed CRISPR/Cas9 miR-873 disrupted SH-SY5Y neuroblastoma cells acquired a neuronal-like morphology and increased expression of ASD important genes ARID1B, SHANK3, ADNP2, ANK2 and CHD8. -

Expression of Periaxin (PRX) Specifically in the Human
www.nature.com/scientificreports OPEN Expression of periaxin (PRX) specifcally in the human cerebrovascular system: PDZ Received: 6 November 2017 Accepted: 13 June 2018 domain-mediated strengthening Published: xx xx xxxx of endothelial barrier function Michael M. Wang1,2,3,4, Xiaojie Zhang1,2, Soo Jung Lee1,2, Snehaa Maripudi1,3, Richard F. Keep2,4,5, Allison M. Johnson4,6,7, Svetlana M. Stamatovic6,7 & Anuska V. Andjelkovic4,6,7 Regulation of cerebral endothelial cell function plays an essential role in changes in blood-brain barrier permeability. Proteins that are important for establishment of endothelial tight junctions have emerged as critical molecules, and PDZ domain containing-molecules are among the most important. We have discovered that the PDZ-domain containing protein periaxin (PRX) is expressed in human cerebral endothelial cells. Surprisingly, PRX protein is not detected in brain endothelium in other mammalian species, suggesting that it could confer human-specifc vascular properties. In endothelial cells, PRX is predominantly localized to the nucleus and not tight junctions. Transcriptome analysis shows that PRX expression suppresses, by at least 50%, a panel of infammatory markers, of which 70% are Type I interferon response genes; only four genes were signifcantly activated by PRX expression. When expressed in mouse endothelial cells, PRX strengthens barrier function, signifcantly increases transendothelial electrical resistance (~35%; p < 0.05), and reduces the permeability of a wide range of molecules. The PDZ domain of PRX is necessary and sufcient for its barrier enhancing properties, since a splice variant (S-PRX) that contains only the PDZ domain, also increases barrier function. PRX also attenuates the permeability enhancing efects of lipopolysaccharide. -

The DLGAP Family: Neuronal Expression, Function and Role in Brain Disorders Andreas H
Rasmussen et al. Molecular Brain (2017) 10:43 DOI 10.1186/s13041-017-0324-9 REVIEW Open Access The DLGAP family: neuronal expression, function and role in brain disorders Andreas H. Rasmussen1, Hanne B. Rasmussen2 and Asli Silahtaroglu1* Abstract The neurotransmitter glutamate facilitates neuronal signalling at excitatory synapses. Glutamate is released from the presynaptic membrane into the synaptic cleft. Across the synaptic cleft glutamate binds to both ion channels and metabotropic glutamate receptors at the postsynapse, which expedite downstream signalling in the neuron. The postsynaptic density, a highly specialized matrix, which is attached to the postsynaptic membrane, controls this downstream signalling. The postsynaptic density also resets the synapse after each synaptic firing. It is composed of numerous proteins including a family of Discs large associated protein 1, 2, 3 and 4 (DLGAP1-4) that act as scaffold proteins in the postsynaptic density. They link the glutamate receptors in the postsynaptic membrane to other glutamate receptors, to signalling proteins and to components of the cytoskeleton. With the central localisation in the postsynapse, the DLGAP family seems to play a vital role in synaptic scaling by regulating the turnover of both ionotropic and metabotropic glutamate receptors in response to synaptic activity. DLGAP family has been directly linked to a variety of psychological and neurological disorders. In this review we focus on the direct and indirect role of DLGAP family on schizophrenia as well as other brain diseases. Keywords: DLGAP1, DLGAP2, DLGAP3, DLGAP4, SAPAP, PSD, GKAP, Schizophrenia, Scaffold proteins, Synaptic scaling Introduction interaction partners, DLGAP1–4 proteins are likely to play The postsynaptic density (PSD) is a highly specialized a role in multiple processes of the PSD. -

An Integrative Analysis of Non-Coding Regulatory DNA Variations Associated with Autism Spectrum Disorder
Molecular Psychiatry (2019) 24:1707–1719 https://doi.org/10.1038/s41380-018-0049-x ARTICLE An integrative analysis of non-coding regulatory DNA variations associated with autism spectrum disorder 1,2 2,3,4 2 2 1 Sarah M. Williams ● Joon Yong An ● Janette Edson ● Michelle Watts ● Valentine Murigneux ● 5,6 7 8 1 Andrew J. O. Whitehouse ● Colin J. Jackson ● Mark A. Bellgrove ● Alexandre S. Cristino ● Charles Claudianos 2,9 Received: 15 October 2016 / Revised: 16 January 2018 / Accepted: 19 February 2018 / Published online: 27 April 2018 © The Author(s) 2018. This article is published with open access Abstract A number of genetic studies have identified rare protein-coding DNA variations associated with autism spectrum disorder (ASD), a neurodevelopmental disorder with significant genetic etiology and heterogeneity. In contrast, the contributions of functional, regulatory genetic variations that occur in the extensive non-protein-coding regions of the genome remain poorly understood. Here we developed a genome-wide analysis to identify the rare single nucleotide variants (SNVs) that occur in non-coding regions and determined the regulatory function and evolutionary conservation of these variants. Using publicly fi 1234567890();,: 1234567890();,: available datasets and computational predictions, we identi ed SNVs within putative regulatory regions in promoters, transcription factor binding sites, and microRNA genes and their target sites. Overall, we found that the regulatory variants in ASD cases were enriched in ASD-risk genes and genes involved in fetal neurodevelopment. As with previously reported coding mutations, we found an enrichment of the regulatory variants associated with dysregulation of neurodevelopmental and synaptic signaling pathways. -
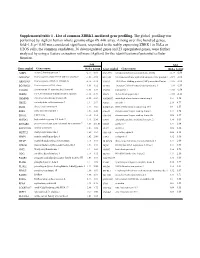
Supplemental Table 1 . List of Common ZBRK1-Mediated Gene Profiling
Supplemental table 1 . List of common ZBRK1-mediated gene profiling. The global profiling was performed by Agilent human whole genome oligo 4X 44K array. Among over five hundred genes, fold>1.5; p< 0.05 was considered significant, responded to the stably expressing ZBRK1 in HeLa or U2OS cells, the common candidates, 36 downregulated genes and 23 upregulated genes, were further analyzed by using a feature extraction software (Agilent) for the identification of potential cellular function. fold fold Gene symbol Genes name HeLa U2OS Gene symbol Genes name HeLa U2OS A2BP1 ataxin 2-binding protein 1 -2.57 -1.61 SNAP25 synaptosomal-associated protein, 25kDa -2.73 -2.50 AF131762 Homo sapiens clone 25218 mRNA sequence -1.82 -1.92 STEAP1 Six transmembrane epithelial antigen of the prostate 1 -2.19 -1.66 AK001903 Homo sapiens cDNA FLJ11041 fis -2.16 -1.72 TAF15 TATA box binding protein (TBP)-associated factor -1.55 -1.61 BG686022 Homo sapiens cDNA clone -3.41 -1.52 TCOF1 Treacher Collins-Franceschetti syndrome 1 -1.59 -1.59 C11orf46 chromosome 11 open reading frame 46 -1.61 -1.59 TNPO1 transportin 1 -1.88 -1.54 CEBPE CCAAT/enhancer binding protein, epsilon -1.52 -1.79 XBP1 X-box binding protein 1 -1.89 -2.11 CHMP4B chromatin modifying protein 4B -3.28 -1.84 AMDHD2 amidohydrolase domain containing 2 1.51 1.54 CHST2 carbohydrate sulfotransferase 2 -1.7 -2.17 ANK1 ankyrin 1 2.19 4.77 DLG4 discs, large homolog 4 -1.77 -1.85 BTBD14A BTB (POZ) domain containing 14A 1.9 1.87 EGR1 early growth response 1 -1.65 -2.22 C9orf5 chromosome 9 open reading frame -

Epigenome-450K-Wide Methylation Signatures of Active Cigarette Smoking: the Young Finns Study
Bioscience Reports (2020) 40 BSR20200596 https://doi.org/10.1042/BSR20200596 Research Article Epigenome-450K-wide methylation signatures of active cigarette smoking: The Young Finns Study Pashupati P. Mishra1,2,3,*, Ismo Hanninen¨ 1,2,3,*, Emma Raitoharju1,2,3, Saara Marttila1,2,3,4, Binisha H. Mishra1,2,3, Nina Mononen1,2,3,MikaKah¨ onen¨ 2,5, Mikko Hurme4,6, Olli Raitakari7,8,9, Petri Tor¨ onen¨ 10, Liisa Holm10,11 and Terho Lehtimaki¨ 1,2,3 Downloaded from http://portlandpress.com/bioscirep/article-pdf/40/7/BSR20200596/887717/bsr-2020-0596.pdf by guest on 27 September 2021 1Department of Clinical Chemistry, Faculty of Medicine and Health Technology, Tampere University, Tampere, Finland; 2Finnish Cardiovascular Research Center-Tampere, Faculty of Medicine and Health Technology, Tampere University, Tampere, Finland; 3Department of Clinical Chemistry, Fimlab Laboratories, Tampere, Finland; 4Gerontology Research Center (GEREC), Tampere University, Tampere, Finland; 5Department of Clinical Physiology, Tampere University Hospital, Tampere, Finland; 6Department of Microbiology and Immunology, Faculty of Medicine and Health Technology, Tampere University, Tampere, Finland; 7Centre for Population Health Research, University of Turku and Turku University Hospital, Turku, Finland; 8Research Centre of Applied and Preventive Cardiovascular Medicine, University of Turku, Turku, Finland; 9Department of Clinical Physiology and Nuclear Medicine, Turku University Hospital, Turku, Finland; 10Institute of Biotechnology, Helsinki Institute of Life Sciences (HiLife), University of Helsinki, Helsinki, Finland; 11Organismal and Evolutionary Biology Research Program, Faculty of Biological and Environmental Sciences, University of Helsinki, Helsinki, Finland Correspondence: Pashupati P. Mishra (pashupati.mishra@tuni.fi) Smoking as a major risk factor for morbidity affects numerous regulatory systems of the human body including DNA methylation. -

And Cell-Type-Specific Transcriptome Is Associated with Cell
OPEN FUS-regulated region- and SUBJECT AREAS: cell-type-specific transcriptome is DEMENTIA CELLULAR NEUROSCIENCE associated with cell selectivity in ALS/ MOTOR NEURON DISEASE ALTERNATIVE SPLICING FTLD Yusuke Fujioka1, Shinsuke Ishigaki1, Akio Masuda2, Yohei Iguchi1, Tsuyoshi Udagawa1, Received Hirohisa Watanabe1, Masahisa Katsuno1, Kinji Ohno2 & Gen Sobue1 27 March 2013 Accepted 1Department of Neurology, 2Division of Neurogenetics, Center for Neurological Diseases and Cancer, Nagoya University 24 July 2013 Graduate School of Medicine, Nagoya, Japan. Published 8 August 2013 FUS is genetically and pathologically linked to amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). To clarify the RNA metabolism cascade regulated by FUS in ALS/FTLD, we compared the FUS-regulated transcriptome profiles in different lineages of primary cells from the central Correspondence and nervous system. The profiles of FUS-mediated gene expression and alternative splicing in motor neurons requests for materials were similar to those of cortical neurons, but not to those in cerebellar neurons despite the similarity of innate transcriptome signature. The gene expression profiles in glial cells were similar to those in motor and should be addressed to cortical neurons. We identified certain neurological diseases-associated genes, including Mapt, Stx1a, and S.I. (ishigaki-ns@umin. Scn8a, among the profiles of gene expression and alternative splicing events regulated by FUS. Thus, net) or G.S. (sobueg@ FUS-regulated transcriptome profiles in each cell-type may determine cellular fate in association with med.nagoya-u.ac.jp) FUS-mediated ALS/FTLD, and identified RNA targets for FUS could be therapeutic targets for ALS/FTLD. myotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by selective motor neuron death in adulthood. -

A High-Density SNP Genome-Wide Linkage Scan in a Large Autism
Molecular Psychiatry (2009) 14, 590–600 & 2009 Nature Publishing Group All rights reserved 1359-4184/09 $32.00 www.nature.com/mp ORIGINAL ARTICLE A high-density SNP genome-wide linkage scan in a large autism extended pedigree K Allen-Brady1, J Miller1, N Matsunami2, J Stevens1, H Block1, M Farley1, L Krasny1, C Pingree1, J Lainhart1, M Leppert1, WM McMahon1 and H Coon1 1Department of Psychiatry, University of Utah, Salt Lake City, UT, USA and 2Department of Human Genetics, University of Utah, Salt Lake City, UT, USA We performed a high-density, single nucleotide polymorphism (SNP), genome-wide scan on a six-generation pedigree from Utah with seven affected males, diagnosed with autism spectrum disorder. Using a two-stage linkage design, we first performed a nonparametric analysis on the entire genome using a 10K SNP chip to identify potential regions of interest. To confirm potentially interesting regions, we eliminated SNPs in high linkage disequilibrium (LD) using a principal components analysis (PCA) method and repeated the linkage results. Three regions met genome-wide significance criteria after controlling for LD: 3q13.2–q13.31 (nonparametric linkage (NPL), 5.58), 3q26.31–q27.3 (NPL, 4.85) and 20q11.21–q13.12 (NPL, 5.56). Two regions met suggestive criteria for significance 7p14.1–p11.22 (NPL, 3.18) and 9p24.3 (NPL, 3.44). All five chromosomal regions are consistent with other published findings. Haplotype sharing results showed that five of the affected subjects shared more than a single chromosomal region of interest with other affected subjects. Although no common autism susceptibility genes were found for all seven autism cases, these results suggest that multiple genetic loci within these regions may contribute to the autism phenotype in this family, and further follow- up of these chromosomal regions is warranted. -

Functional Genomics of Cohesin Acetyltransferases in Human Cells
FUNCTIONAL GENOMICS OF COHESIN ACETYLTRANSFERASES IN HUMAN CELLS by Sadia Rahman A Dissertation Presented to the Faculty of the Louis V. Gerstner, Jr. Graduate School of Biomedical Sciences, Memorial Sloan Kettering Cancer Center in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy New York, NY March, 2014 __________________________ _________________ Prasad V. Jallepalli, MD, PhD Date Dissertation Mentor ABSTRACT Accurate chromosome segregation during cell division requires that sister chromatids be physically linked from the time of their replication until their separation at anaphase. The cohesin complex, consisting of SMC1, SMC3, RAD21 and SCC3 arranges to form a ring-shaped structure that holds sister chromatids together. Acetylation of the cohesin SMC3 subunit by acetyltransferases ESCO1 and ESCO2 is essential for cohesion establishment. In addition to cohesion, cohesin also has roles in gene expression through its regulation of chromatin architecture. Acetylation of cohesin by ESCO1/2 is regulated temporally and spatially. In human cells, it begins in G1 phase, rises in S-phase and persists until mitosis. The reaction occurs only on DNA-bound cohesin and SMC3 is quickly deacetylated after cohesin is removed from DNA. In this study, we map genome-wide ESCO1/2 and AcSMC3 sites by ChIP- Seq, study their regulation, and contribution to cohesion and gene expression functions. Genome-wide mapping of ESCO1/2 reveals that they differ in their distribution: ESCO1 has many discrete binding sites that largely overlap with cohesin/CTCF sites, whereas ESCO2 has few sites of enrichment. A monoclonal antibody against the acetylated form of cohesin was also generated in this study to map cohesin acetylation, and this shows that cohesin is already acetylated in G1 at the majority of its sites and that this depends on ESCO1. -

High-Density Array Comparative Genomic Hybridization Detects Novel Copy Number Alterations in Gastric Adenocarcinoma
ANTICANCER RESEARCH 34: 6405-6416 (2014) High-density Array Comparative Genomic Hybridization Detects Novel Copy Number Alterations in Gastric Adenocarcinoma ALINE DAMASCENO SEABRA1,2*, TAÍSSA MAÍRA THOMAZ ARAÚJO1,2*, FERNANDO AUGUSTO RODRIGUES MELLO JUNIOR1,2, DIEGO DI FELIPE ÁVILA ALCÂNTARA1,2, AMANDA PAIVA DE BARROS1,2, PAULO PIMENTEL DE ASSUMPÇÃO2, RAQUEL CARVALHO MONTENEGRO1,2, ADRIANA COSTA GUIMARÃES1,2, SAMIA DEMACHKI2, ROMMEL MARIO RODRÍGUEZ BURBANO1,2 and ANDRÉ SALIM KHAYAT1,2 1Human Cytogenetics Laboratory and 2Oncology Research Center, Federal University of Pará, Belém Pará, Brazil Abstract. Aim: To investigate frequent quantitative alterations gastric cancer is the second most frequent cancer in men and of intestinal-type gastric adenocarcinoma. Materials and the third in women (4). The state of Pará has a high Methods: We analyzed genome-wide DNA copy numbers of 22 incidence of gastric adenocarcinoma and this disease is a samples and using CytoScan® HD Array. Results: We identified public health problem, since mortality rates are above the 22 gene alterations that to the best of our knowledge have not Brazilian average (5). been described for gastric cancer, including of v-erb-b2 avian This tumor can be classified into two histological types, erythroblastic leukemia viral oncogene homolog 4 (ERBB4), intestinal and diffuse, according to Laurén (4, 6, 7). The SRY (sex determining region Y)-box 6 (SOX6), regulator of intestinal type predominates in high-risk areas, such as telomere elongation helicase 1 (RTEL1) and UDP- Brazil, and arises from precursor lesions, whereas the diffuse Gal:betaGlcNAc beta 1,4- galactosyltransferase, polypeptide 5 type has a similar distribution in high- and low-risk areas and (B4GALT5). -

An Integrative Genomic Study Implicates the Postsynaptic Density in the Pathogenesis of Bipolar Disorder
Neuropsychopharmacology (2016) 41, 886–895 © 2016 American College of Neuropsychopharmacology. All rights reserved 0893-133X/16 www.neuropsychopharmacology.org An Integrative Genomic Study Implicates the Postsynaptic Density in the Pathogenesis of Bipolar Disorder ,1 1,3 2 1 Nirmala Akula* , Jens R Wendland , Kwang H Choi and Francis J McMahon 1 Human Genetics Branch, National Institute of Mental Health Intramural Research Program (NIMH-IRP), National Institutes of Health, US 2 Department of Health and Human Services, Bethesda, MD, USA; Department of Psychiatry, Uniformed Services University of the Health Sciences, Bethesda, MD, USA Genome-wide association studies (GWAS) have identified several common variants associated with bipolar disorder (BD), but the — biological meaning of these findings remains unclear. Integrative genomics the integration of GWAS signals with gene expression — data may illuminate genes and gene networks that have key roles in the pathogenesis of BD. We applied weighted gene co-expression network analysis (WGCNA), which exploits patterns of co-expression among genes, to brain transcriptome data obtained by sequencing of poly-A RNA derived from postmortem dorsolateral prefrontal cortex from people with BD, along with age- and sex-matched controls. WGCNA identified 33 gene modules. Many of the modules corresponded closely to those previously reported in human cortex. Three modules were associated with BD, enriched for genes differentially expressed in BD, and also enriched for signals in prior GWAS of BD. Functional analysis of genes within these modules revealed significant enrichment of several functionally related sets of genes, especially those involved in the postsynaptic density (PSD). These results provide convergent support for the hypothesis that dysregulation of genes involved in the PSD is a key factor in the pathogenesis of BD. -

Integrative Functional Analyses of the Neurodegenerative Disease-Associated TECPR2 Gene Reveal Its Diverse Roles
Integrative functional analyses of the neurodegenerative disease-associated TECPR2 gene reveal its diverse roles Ido Shalev Ben-Gurion University of the Negev Judith Somekh University of Haifa Alal Eran ( [email protected] ) Ben-Gurion University of the Negev https://orcid.org/0000-0001-6784-7597 Research article Keywords: Integrative functional analysis, neurodegenerative disorders, autophagy, ribosome, TECPR2 Posted Date: January 30th, 2020 DOI: https://doi.org/10.21203/rs.2.22274/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract Background Loss of tectonin β-propeller repeat-containing 2 (TECPR2) function has been implicated in an array of neurodegenerative disorders, yet its physiological function remains largely unknown. Understanding TECPR2 function is essential for developing much needed precision therapeutics for TECPR2-related diseases. Methods We leveraged the considerable amounts of functional data to obtain a comprehensive perspective of the role of TECPR2 in health and disease. We integrated expression patterns, population variation, phylogenetic proling, protein-protein interactions, and regulatory network data for a minimally biased multimodal functional analysis. Genes and proteins linked to TECPR2 via multiple lines of evidence were subject to functional enrichment analyses to identify molecular mechanisms involving TECPR2. Results TECPR2 was found to be part of a tight neurodevelopmental gene expression program that includes KIF1A, ATXN1, TOM1L2, and FA2H, all implicated in neurological diseases. Functional enrichment analyses of TECPR2-related genes converged on a role in late autophagy and ribosomal processes. Large-scale population variation data demonstrated that this role is nonredundant. Conclusions TECPR2 might serve as an indicator for the energy balance between protein synthesis and autophagy, and a marker for diseases associated with their imbalance, such as Alzheimer’s disease, Huntington’s disease, and various cancers.