An Integrative Genomic Study Implicates the Postsynaptic Density in the Pathogenesis of Bipolar Disorder
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deregulated Gene Expression Pathways in Myelodysplastic Syndrome Hematopoietic Stem Cells
Leukemia (2010) 24, 756–764 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00 www.nature.com/leu ORIGINAL ARTICLE Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells A Pellagatti1, M Cazzola2, A Giagounidis3, J Perry1, L Malcovati2, MG Della Porta2,MJa¨dersten4, S Killick5, A Verma6, CJ Norbury7, E Hellstro¨m-Lindberg4, JS Wainscoat1 and J Boultwood1 1LRF Molecular Haematology Unit, NDCLS, John Radcliffe Hospital, Oxford, UK; 2Department of Hematology Oncology, University of Pavia Medical School, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; 3Medizinische Klinik II, St Johannes Hospital, Duisburg, Germany; 4Division of Hematology, Department of Medicine, Karolinska Institutet, Stockholm, Sweden; 5Department of Haematology, Royal Bournemouth Hospital, Bournemouth, UK; 6Albert Einstein College of Medicine, Bronx, NY, USA and 7Sir William Dunn School of Pathology, University of Oxford, Oxford, UK To gain insight into the molecular pathogenesis of the the World Health Organization.6,7 Patients with refractory myelodysplastic syndromes (MDS), we performed global gene anemia (RA) with or without ringed sideroblasts, according to expression profiling and pathway analysis on the hemato- poietic stem cells (HSC) of 183 MDS patients as compared with the the French–American–British classification, were subdivided HSC of 17 healthy controls. The most significantly deregulated based on the presence or absence of multilineage dysplasia. In pathways in MDS include interferon signaling, thrombopoietin addition, patients with RA with excess blasts (RAEB) were signaling and the Wnt pathways. Among the most signifi- subdivided into two categories, RAEB1 and RAEB2, based on the cantly deregulated gene pathways in early MDS are immuno- percentage of bone marrow blasts. -

Molecular Associations and Clinical Significance of Raps In
ORIGINAL RESEARCH published: 21 June 2021 doi: 10.3389/fmolb.2021.677979 Molecular Associations and Clinical Significance of RAPs in Hepatocellular Carcinoma Sarita Kumari 1†, Mohit Arora 2†, Jay Singh 1, Lokesh K. Kadian 2, Rajni Yadav 3, Shyam S. Chauhan 2* and Anita Chopra 1* 1Laboratory Oncology Unit, Dr. BRA-IRCH, All India Institute of Medical Sciences, New Delhi, India, 2Department of Biochemistry, All India Institute of Medical Sciences, New Delhi, India, 3Department of Pathology, All India Institute of Medical Sciences, New Delhi, India Hepatocellular carcinoma (HCC) is an aggressive gastrointestinal malignancy with a high rate of mortality. Multiple studies have individually recognized members of RAP gene family as critical regulators of tumor progression in several cancers, including hepatocellular carcinoma. These studies suffer numerous limitations including a small sample size and lack of analysis of various clinicopathological and molecular Edited by: Veronica Aran, features. In the current study, we utilized authoritative multi-omics databases to Instituto Estadual do Cérebro Paulo determine the association of RAP gene family expression and detailed molecular and Niemeyer (IECPN), Brazil clinicopathological features in hepatocellular carcinoma (HCC). All five RAP genes Reviewed by: were observed to harbor dysregulated expression in HCC compared to normal liver Pooja Panwalkar, Weill Cornell Medicine, United States tissues. RAP2A exhibited strongest ability to differentiate tumors from the normal Jasminka Omerovic, tissues. RAP2A expression was associated with progressive tumor grade, TP53 and University of Split, Croatia CTNNB1 mutation status. Additionally, RAP2A expression was associated with the *Correspondence: Anita Chopra alteration of its copy numbers and DNA methylation. RAP2A also emerged as an [email protected] independent marker for patient prognosis. -

Autism-Associated Mir-873 Regulates ARID1B, SHANK3 and NRXN2
Lu et al. Translational Psychiatry (2020) 10:418 https://doi.org/10.1038/s41398-020-01106-8 Translational Psychiatry ARTICLE Open Access Autism-associated miR-873 regulates ARID1B, SHANK3 and NRXN2 involved in neurodevelopment Jing Lu 1, Yan Zhu 1, Sarah Williams2, Michelle Watts 3,MaryA.Tonta1, Harold A. Coleman1, Helena C. Parkington1 and Charles Claudianos 1,4 Abstract Autism spectrum disorders (ASD) are highly heritable neurodevelopmental disorders with significant genetic heterogeneity. Noncoding microRNAs (miRNAs) are recognised as playing key roles in development of ASD albeit the function of these regulatory genes remains unclear. We previously conducted whole-exome sequencing of Australian families with ASD and identified four novel single nucleotide variations in mature miRNA sequences. A pull-down transcriptome analysis using transfected SH-SY5Y cells proposed a mechanistic model to examine changes in binding affinity associated with a unique mutation found in the conserved ‘seed’ region of miR-873-5p (rs777143952: T > A). Results suggested several ASD-risk genes were differentially targeted by wild-type and mutant miR-873 variants. In the current study, a dual-luciferase reporter assay confirmed miR-873 variants have a 20-30% inhibition/dysregulation effect on candidate autism risk genes ARID1B, SHANK3 and NRXN2 and also confirmed the affected expression with qPCR. In vitro mouse hippocampal neurons transfected with mutant miR-873 showed less morphological complexity and enhanced sodium currents and excitatory neurotransmission compared to cells transfected with wild-type miR- 873. A second in vitro study showed CRISPR/Cas9 miR-873 disrupted SH-SY5Y neuroblastoma cells acquired a neuronal-like morphology and increased expression of ASD important genes ARID1B, SHANK3, ADNP2, ANK2 and CHD8. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Examination of the Expression and Prognostic Significance of Dlgaps in Gastric Cancer Using the TCGA Database and Bioinformatic Analysis
MOLECULAR MEDICINE REPORTS 18: 5621-5629, 2018 Examination of the expression and prognostic significance of DLGAPs in gastric cancer using the TCGA database and bioinformatic analysis JIAXIN LIU1, ZHAO LIU2, XIAOZHI ZHANG3, TUOTUO GONG3 and DEMAO YAO1 Departments of 1Gerontological Surgery, 2Oncological Surgery and 3Radiation Oncology, First Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi 710061, P.R. China Received February 3, 2018; Accepted September 21, 2018 DOI: 10.3892/mmr.2018.9574 Abstract. The discs large-associated protein (DLGAP) Introduction family has been implicated in psychological and neurological diseases. However, few studies have explored the association Gastric cancer (GC) is a highly aggressive disease, with an inci- between the expression of DLGAPs and different types of dence that is ranked fifth among all types of cancers worldwide cancer. Therefore, the present study analyzed the status of in recent years (1,2). There are nearly 1,000,000 new cases of DLGAPs in gastric cancer (GC) using bioinformatic tools. GC diagnosed globally each year (3). The majority of patients Analyses of data obtained from The Cancer Genome Atlas and succumb to GC due to late recurrence or distant metastasis, and Gene Expression Omnibus databases revealed that there was the 5-year survival rate for GC patients is <5% (4). Thus, there selective upregulation of DLGAP4 and DLGAP5 expression is an urgent requirement to identify biomarkers associated with in GC tissues when compared with normal gastric tissues. the prognosis of patients with GC. Although some targets are In addition, survival analysis using OncoLnc indicated that currently in clinical use, including human epidermal growth high expression of DLGAP4 was significantly correlated factor receptor 2 (5,6), vascular endothelial growth factor with shorter overall survival for all GC patients. -

Expression of Periaxin (PRX) Specifically in the Human
www.nature.com/scientificreports OPEN Expression of periaxin (PRX) specifcally in the human cerebrovascular system: PDZ Received: 6 November 2017 Accepted: 13 June 2018 domain-mediated strengthening Published: xx xx xxxx of endothelial barrier function Michael M. Wang1,2,3,4, Xiaojie Zhang1,2, Soo Jung Lee1,2, Snehaa Maripudi1,3, Richard F. Keep2,4,5, Allison M. Johnson4,6,7, Svetlana M. Stamatovic6,7 & Anuska V. Andjelkovic4,6,7 Regulation of cerebral endothelial cell function plays an essential role in changes in blood-brain barrier permeability. Proteins that are important for establishment of endothelial tight junctions have emerged as critical molecules, and PDZ domain containing-molecules are among the most important. We have discovered that the PDZ-domain containing protein periaxin (PRX) is expressed in human cerebral endothelial cells. Surprisingly, PRX protein is not detected in brain endothelium in other mammalian species, suggesting that it could confer human-specifc vascular properties. In endothelial cells, PRX is predominantly localized to the nucleus and not tight junctions. Transcriptome analysis shows that PRX expression suppresses, by at least 50%, a panel of infammatory markers, of which 70% are Type I interferon response genes; only four genes were signifcantly activated by PRX expression. When expressed in mouse endothelial cells, PRX strengthens barrier function, signifcantly increases transendothelial electrical resistance (~35%; p < 0.05), and reduces the permeability of a wide range of molecules. The PDZ domain of PRX is necessary and sufcient for its barrier enhancing properties, since a splice variant (S-PRX) that contains only the PDZ domain, also increases barrier function. PRX also attenuates the permeability enhancing efects of lipopolysaccharide. -

The DLGAP Family: Neuronal Expression, Function and Role in Brain Disorders Andreas H
Rasmussen et al. Molecular Brain (2017) 10:43 DOI 10.1186/s13041-017-0324-9 REVIEW Open Access The DLGAP family: neuronal expression, function and role in brain disorders Andreas H. Rasmussen1, Hanne B. Rasmussen2 and Asli Silahtaroglu1* Abstract The neurotransmitter glutamate facilitates neuronal signalling at excitatory synapses. Glutamate is released from the presynaptic membrane into the synaptic cleft. Across the synaptic cleft glutamate binds to both ion channels and metabotropic glutamate receptors at the postsynapse, which expedite downstream signalling in the neuron. The postsynaptic density, a highly specialized matrix, which is attached to the postsynaptic membrane, controls this downstream signalling. The postsynaptic density also resets the synapse after each synaptic firing. It is composed of numerous proteins including a family of Discs large associated protein 1, 2, 3 and 4 (DLGAP1-4) that act as scaffold proteins in the postsynaptic density. They link the glutamate receptors in the postsynaptic membrane to other glutamate receptors, to signalling proteins and to components of the cytoskeleton. With the central localisation in the postsynapse, the DLGAP family seems to play a vital role in synaptic scaling by regulating the turnover of both ionotropic and metabotropic glutamate receptors in response to synaptic activity. DLGAP family has been directly linked to a variety of psychological and neurological disorders. In this review we focus on the direct and indirect role of DLGAP family on schizophrenia as well as other brain diseases. Keywords: DLGAP1, DLGAP2, DLGAP3, DLGAP4, SAPAP, PSD, GKAP, Schizophrenia, Scaffold proteins, Synaptic scaling Introduction interaction partners, DLGAP1–4 proteins are likely to play The postsynaptic density (PSD) is a highly specialized a role in multiple processes of the PSD. -

Genome-Wide Association Study of Brain Amyloid Deposition As Measured by Pittsburgh Compound-B (Pib)-PET Imaging
Molecular Psychiatry (2021) 26:309–321 https://doi.org/10.1038/s41380-018-0246-7 ARTICLE Genome-wide association study of brain amyloid deposition as measured by Pittsburgh Compound-B (PiB)-PET imaging 1,2 3,4 5 1 3,4 1 Qi Yan ● Kwangsik Nho ● Jorge L. Del-Aguila ● Xingbin Wang ● Shannon L. Risacher ● Kang-Hsien Fan ● 6,7 8 7,9 6,7,8 1 Beth E. Snitz ● Howard J. Aizenstein ● Chester A. Mathis ● Oscar L. Lopez ● F. Yesim Demirci ● 1 6,7,8 3,4 Eleanor Feingold ● William E. Klunk ● Andrew J. Saykin ● for the Alzheimer’s Disease Neuroimaging 5 1,7,8 Initiative (ADNI) ● Carlos Cruchaga ● M. Ilyas Kamboh Received: 1 December 2017 / Accepted: 31 July 2018 / Published online: 25 October 2018 © The Author(s) 2018. This article is published with open access Abstract Deposition of amyloid plaques in the brain is one of the two main pathological hallmarks of Alzheimer’s disease (AD). Amyloid positron emission tomography (PET) is a neuroimaging tool that selectively detects in vivo amyloid deposition in the brain and is a reliable endophenotype for AD that complements cerebrospinal fluid biomarkers with regional information. We measured in vivo amyloid deposition in the brains of ~1000 subjects from three collaborative AD centers and ADNI 11 1234567890();,: 1234567890();,: using C-labeled Pittsburgh Compound-B (PiB)-PET imaging followed by meta-analysis of genome-wide association studies, first to our knowledge for PiB-PET, to identify novel genetic loci for this endophenotype. The APOE region showed the most significant association where several SNPs surpassed the genome-wide significant threshold, with APOE*4 being most significant (P-meta = 9.09E-30; β = 0.18). -

An Integrative Analysis of Non-Coding Regulatory DNA Variations Associated with Autism Spectrum Disorder
Molecular Psychiatry (2019) 24:1707–1719 https://doi.org/10.1038/s41380-018-0049-x ARTICLE An integrative analysis of non-coding regulatory DNA variations associated with autism spectrum disorder 1,2 2,3,4 2 2 1 Sarah M. Williams ● Joon Yong An ● Janette Edson ● Michelle Watts ● Valentine Murigneux ● 5,6 7 8 1 Andrew J. O. Whitehouse ● Colin J. Jackson ● Mark A. Bellgrove ● Alexandre S. Cristino ● Charles Claudianos 2,9 Received: 15 October 2016 / Revised: 16 January 2018 / Accepted: 19 February 2018 / Published online: 27 April 2018 © The Author(s) 2018. This article is published with open access Abstract A number of genetic studies have identified rare protein-coding DNA variations associated with autism spectrum disorder (ASD), a neurodevelopmental disorder with significant genetic etiology and heterogeneity. In contrast, the contributions of functional, regulatory genetic variations that occur in the extensive non-protein-coding regions of the genome remain poorly understood. Here we developed a genome-wide analysis to identify the rare single nucleotide variants (SNVs) that occur in non-coding regions and determined the regulatory function and evolutionary conservation of these variants. Using publicly fi 1234567890();,: 1234567890();,: available datasets and computational predictions, we identi ed SNVs within putative regulatory regions in promoters, transcription factor binding sites, and microRNA genes and their target sites. Overall, we found that the regulatory variants in ASD cases were enriched in ASD-risk genes and genes involved in fetal neurodevelopment. As with previously reported coding mutations, we found an enrichment of the regulatory variants associated with dysregulation of neurodevelopmental and synaptic signaling pathways. -

New Secreted Toxins and Immunity Proteins Encoded Within the Type VI Secretion System Gene Cluster of Serratia Marcescens
Molecular Microbiology (2012) 86(4), 921–936 doi:10.1111/mmi.12028 First published online 27 September 2012 New secreted toxins and immunity proteins encoded within the Type VI secretion system gene cluster of Serratia marcescens Grant English,1† Katharina Trunk,1† into how pathogens utilize antibacterial T6SSs to Vincenzo A. Rao,2 Velupillai Srikannathasan,2 overcome competitors and succeed in polymicrobial William N. Hunter2 and Sarah J. Coulthurst1* niches. 1Division of Molecular Microbiology, College of Life Sciences, University of Dundee, Dundee, UK. 2Division of Biological Chemistry and Drug Discovery, Introduction College of Life Sciences, University of Dundee, Dundee, Protein secretion systems and their substrates are central UK. to bacterial virulence and interaction with other organisms (Gerlach and Hensel, 2007). Six different secretion Summary systems (Types I–VI) are used by Gram-negative bacteria to transport specific proteins to the exterior of the bacterial Protein secretion systems are critical to bacterial viru- cell or further inject them into target cells. The most lence and interactions with other organisms. The Type recently described of these is the Type VI secretion VI secretion system (T6SS) is found in many bacterial system (T6SS) (Filloux et al., 2008). T6SSs are complex species and is used to target either eukaryotic cells or multi-protein assemblies that span both bacterial mem- competitor bacteria. However, T6SS-secreted proteins branes and inject effector proteins directly from the bac- have proven surprisingly elusive. Here, we identified terial cytoplasm into target cells (Bonemann et al., 2010; two secreted substrates of the antibacterial T6SS from Cascales and Cambillau, 2012). T6SSs are encoded by the opportunistic human pathogen, Serratia marces- large, variable gene clusters that contain 13 ‘core’ essen- cens. -
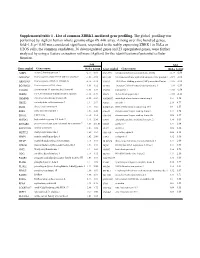
Supplemental Table 1 . List of Common ZBRK1-Mediated Gene Profiling
Supplemental table 1 . List of common ZBRK1-mediated gene profiling. The global profiling was performed by Agilent human whole genome oligo 4X 44K array. Among over five hundred genes, fold>1.5; p< 0.05 was considered significant, responded to the stably expressing ZBRK1 in HeLa or U2OS cells, the common candidates, 36 downregulated genes and 23 upregulated genes, were further analyzed by using a feature extraction software (Agilent) for the identification of potential cellular function. fold fold Gene symbol Genes name HeLa U2OS Gene symbol Genes name HeLa U2OS A2BP1 ataxin 2-binding protein 1 -2.57 -1.61 SNAP25 synaptosomal-associated protein, 25kDa -2.73 -2.50 AF131762 Homo sapiens clone 25218 mRNA sequence -1.82 -1.92 STEAP1 Six transmembrane epithelial antigen of the prostate 1 -2.19 -1.66 AK001903 Homo sapiens cDNA FLJ11041 fis -2.16 -1.72 TAF15 TATA box binding protein (TBP)-associated factor -1.55 -1.61 BG686022 Homo sapiens cDNA clone -3.41 -1.52 TCOF1 Treacher Collins-Franceschetti syndrome 1 -1.59 -1.59 C11orf46 chromosome 11 open reading frame 46 -1.61 -1.59 TNPO1 transportin 1 -1.88 -1.54 CEBPE CCAAT/enhancer binding protein, epsilon -1.52 -1.79 XBP1 X-box binding protein 1 -1.89 -2.11 CHMP4B chromatin modifying protein 4B -3.28 -1.84 AMDHD2 amidohydrolase domain containing 2 1.51 1.54 CHST2 carbohydrate sulfotransferase 2 -1.7 -2.17 ANK1 ankyrin 1 2.19 4.77 DLG4 discs, large homolog 4 -1.77 -1.85 BTBD14A BTB (POZ) domain containing 14A 1.9 1.87 EGR1 early growth response 1 -1.65 -2.22 C9orf5 chromosome 9 open reading frame -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV).