Consensus Transcriptional Regulatory Networks of Coronavirus-Infected
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

The T-ALL Related Gene BCL11B Regulates the Initial Stages of Human T-Cell Differentiation
Leukemia (2017) 31, 2503–2514 © 2017 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 0887-6924/17 www.nature.com/leu ORIGINAL ARTICLE The T-ALL related gene BCL11B regulates the initial stages of human T-cell differentiation VL Ha1, A Luong1,FLi2, D Casero3, J Malvar1,YMKim1,4, R Bhatia5, GM Crooks3,6,7,8 and C Parekh1,4 The initial stages of T-cell differentiation are characterized by a progressive commitment to the T-cell lineage, a process that involves the loss of alternative (myelo-erythroid, NK, B) lineage potentials. Aberrant differentiation during these stages can result in T-cell acute lymphoblastic leukemia (T-ALL). However, the mechanisms regulating the initial stages of human T-cell differentiation are obscure. Through loss of function studies, we showed BCL11B, a transcription factor recurrently mutated T-ALL, is essential for T-lineage commitment, particularly the repression of NK and myeloid potentials, and the induction of T-lineage genes, during the initial stages of human T-cell differentiation. In gain of function studies, BCL11B inhibited growth of and induced a T-lineage transcriptional program in T-ALL cells. We found previously unknown differentiation stage-specific DNA binding of BCL11B at multiple T-lineage genes; target genes showed BCL11B-dependent expression, suggesting a transcriptional activator role for BCL11B at these genes. Transcriptional analyses revealed differences in the regulatory actions of BCL11B between human and murine thymopoiesis. Our studies show BCL11B is a key regulator of the initial stages of human T-cell differentiation and delineate the BCL11B transcriptional program, enabling the dissection of the underpinnings of normal T-cell differentiation and providing a resource for understanding dysregulations in T-ALL. -

Core Transcriptional Regulatory Circuitries in Cancer
Oncogene (2020) 39:6633–6646 https://doi.org/10.1038/s41388-020-01459-w REVIEW ARTICLE Core transcriptional regulatory circuitries in cancer 1 1,2,3 1 2 1,4,5 Ye Chen ● Liang Xu ● Ruby Yu-Tong Lin ● Markus Müschen ● H. Phillip Koeffler Received: 14 June 2020 / Revised: 30 August 2020 / Accepted: 4 September 2020 / Published online: 17 September 2020 © The Author(s) 2020. This article is published with open access Abstract Transcription factors (TFs) coordinate the on-and-off states of gene expression typically in a combinatorial fashion. Studies from embryonic stem cells and other cell types have revealed that a clique of self-regulated core TFs control cell identity and cell state. These core TFs form interconnected feed-forward transcriptional loops to establish and reinforce the cell-type- specific gene-expression program; the ensemble of core TFs and their regulatory loops constitutes core transcriptional regulatory circuitry (CRC). Here, we summarize recent progress in computational reconstitution and biologic exploration of CRCs across various human malignancies, and consolidate the strategy and methodology for CRC discovery. We also discuss the genetic basis and therapeutic vulnerability of CRC, and highlight new frontiers and future efforts for the study of CRC in cancer. Knowledge of CRC in cancer is fundamental to understanding cancer-specific transcriptional addiction, and should provide important insight to both pathobiology and therapeutics. 1234567890();,: 1234567890();,: Introduction genes. Till now, one critical goal in biology remains to understand the composition and hierarchy of transcriptional Transcriptional regulation is one of the fundamental mole- regulatory network in each specified cell type/lineage. -

RNA-Binding Protein Hnrnpll Regulates Mrna Splicing and Stability During B-Cell to Plasma-Cell Differentiation
RNA-binding protein hnRNPLL regulates mRNA splicing and stability during B-cell to plasma-cell differentiation Xing Changa,b, Bin Lic, and Anjana Raoa,b,d,e,1 Divisions of aSignaling and Gene Expression and cVaccine Discovery, La Jolla Institute for Allergy and Immunology, La Jolla, CA 92037; bSanford Consortium for Regenerative Medicine, La Jolla, CA 92037; and dDepartment of Pharmacology and eMoores Cancer Center, University of California at San Diego, La Jolla, CA 92093 Contributed by Anjana Rao, December 2, 2014 (sent for review July 20, 2014) Posttranscriptional regulation is a major mechanism to rewire the RBP-binding sites, thus validating the specificity of RBP binding transcriptomes during differentiation. Heterogeneous nuclear to coprecipitating RNAs and mapping RBP-binding sites on the RNA-binding protein LL (hnRNPLL) is specifically induced in terminally validated RNAs at close to single-nucleotide resolution (8). differentiated lymphocytes, including effector T cells and plasma Heterogeneous nuclear RNA-binding proteins (hnRNPs) is cells. To study the molecular functions of hnRNPLL at a genome- the term applied to a collection of unrelated nuclear RBPs. wide level, we identified hnRNPLL RNA targets and binding sites in hnRNPLL was identified through a targeted lentiviral shRNA plasma cells through integrated Photoactivatable-Ribonucleoside- screen for regulators of CD45RA to CD45RO switching during Enhanced Cross-Linking and Immunoprecipitation (PAR-CLIP) and memory T-cell development (9) and independently through RNA sequencing. hnRNPLL preferentially recognizes CA dinucleo- two separate screens performed by different groups for exclusion tide-containing sequences in introns and 3′ untranslated regions of CD45 exon 4 in a minigene context (10) and for altered CD44 (UTRs), promotes exon inclusion or exclusion in a context-dependent and CD45R expression on T cells in N-ethyl-N-nitrosourea manner, and stabilizes mRNA when associated with 3′ UTRs. -
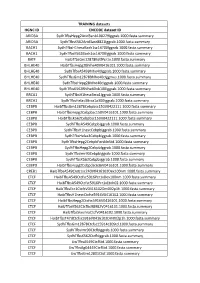
TRAINING Datasets HGNC ID ENCODE Dataset ID ARID3A
TRAINING datasets HGNC ID ENCODE dataset ID ARID3A SydhT+sHepg2Arid3anb100279Iggrab.1000.fasta.summary ARID3A SydhT+sK562Arid3asC8821Iggrab.1000.fasta.summary BACH1 SydhT+sH1hesCBaCh1sC14700Iggrab.1000.fasta.summary BACH1 SydhT+sK562BaCh1sC14700Iggrab.1000.fasta.summary BATF HaibT+sGm12878BaJPCr1x.1000.fasta.summary BHLHE40 HaibT+sHepg2Bhlhe40V0416101.1000.fasta.summary BHLHE40 SydhT+sA549Bhlhe40Iggrab.1000.fasta.summary BHLHE40 SydhT+sGm12878Bhlhe40CIggmus.1000.fasta.summary BHLHE40 SydhT+sHepg2Bhlhe40CIggrab.1000.fasta.summary BHLHE40 SydhT+sK562Bhlhe40nb100Iggrab.1000.fasta.summary BRCA1 SydhT+sH1hesCBrCa1Iggrab.1000.fasta.summary BRCA1 SydhT+sHelas3BrCa1a300Iggrab.1000.fasta.summary CEBPB HaibT+sGm12878CebpbsC150V0422111.1000.fasta.summary CEBPB HaibT+sHepg2CebpbsC150V0416101.1000.fasta.summary CEBPB HaibT+sK562CebpbsC150V0422111.1000.fasta.summary CEBPB SydhT+sA549CebpbIggrab.1000.fasta.summary CEBPB SydhT+sH1hesCCebpbIggrab.1000.fasta.summary CEBPB SydhT+sHelas3CebpbIggrab.1000.fasta.summary CEBPB SydhT+sHepg2CebpbForsklnStd.1000.fasta.summary CEBPB SydhT+sHepg2CebpbIggrab.1000.fasta.summary CEBPB SydhT+sImr90CebpbIggrab.1000.fasta.summary CEBPB SydhT+sK562CebpbIggrab.1000.fasta.summary CEBPD HaibT+sHepg2CebpdsC636V0416101.1000.fasta.summary CREB1 HaibT+sA549Creb1sC240V0416102Dex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xDex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xEtoh02.1000.fasta.summary CTCF HaibT+sECC1CtCfCV0416102Dm002p1h.1000.fasta.summary CTCF HaibT+sH1hesCCtCfsC5916V0416102.1000.fasta.summary -

G2 Phase Cell Cycle Regulation by E2F4 Following Genotoxic Stress
G2 Phase Cell Cycle Regulation by E2F4 Following Genotoxic Stress by MEREDITH ELLEN CROSBY Submitted in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Thesis Advisor: Dr. Alex Almasan Department of Environmental Health Sciences CASE WESTERN RESERVE UNIVERSITY May, 2006 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the dissertation of ______________________________________________________ candidate for the Ph.D. degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. TABLE OF CONTENTS TABLE OF CONTENTS………………………………………………………………….1 LIST OF FIGURES……………………………………………………………………….5 LIST OF TABLES………………………………………………………………………...7 ACKNOWLEDGEMENTS……………………………………………………………….8 LIST OF ABBREVIATIONS……………………………………………………………10 ABSTRACT……………………………………………………………………………...15 CHAPTER 1. INTRODUCTION 1.1. CELL CYCLE REGULATION: HISTORICAL OVERVIEW………………...17 1.2. THE E2F FAMILY OF TRANSCRIPTION FACTORS……………………….22 1.3. E2F AND CELL CYCLE CONTROL 1.3.1. G0/G1 Phase Transition………………………………………………….28 1.3.2. S Phase…………………………………………………………………...28 1.3.3. G2/M Phase Transition…………………………………………………..30 1.4. -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

2017.08.28 Anne Barry-Reidy Thesis Final.Pdf
REGULATION OF BOVINE β-DEFENSIN EXPRESSION THIS THESIS IS SUBMITTED TO THE UNIVERSITY OF DUBLIN FOR THE DEGREE OF DOCTOR OF PHILOSOPHY 2017 ANNE BARRY-REIDY SCHOOL OF BIOCHEMISTRY & IMMUNOLOGY TRINITY COLLEGE DUBLIN SUPERVISORS: PROF. CLIONA O’FARRELLY & DR. KIERAN MEADE TABLE OF CONTENTS DECLARATION ................................................................................................................................. vii ACKNOWLEDGEMENTS ................................................................................................................... viii ABBREVIATIONS ................................................................................................................................ix LIST OF FIGURES............................................................................................................................. xiii LIST OF TABLES .............................................................................................................................. xvii ABSTRACT ........................................................................................................................................xix Chapter 1 Introduction ........................................................................................................ 1 1.1 Antimicrobial/Host-defence peptides ..................................................................... 1 1.2 Defensins................................................................................................................. 1 1.3 β-defensins ............................................................................................................. -

The Histone Methyltransferase DOT1L Prevents Antigen-Independent
bioRxiv preprint doi: https://doi.org/10.1101/826255; this version posted November 18, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The histone methyltransferase DOT1L prevents antigen-independent differentiation and safeguards epigenetic identity of CD8+ T cells Eliza Mari Kwesi-Maliepaard1*, Muhammad Assad Aslam2,3*, Mir Farshid Alemdehy2*, Teun van den Brand4, Chelsea McLean1, Hanneke Vlaming1, Tibor van Welsem1, Tessy Korthout1, Cesare Lancini1, Sjoerd Hendriks1, Tomasz Ahrends5, Dieke van Dinther6, Joke M.M. den Haan6, Jannie Borst5, Elzo de Wit4, Fred van Leeuwen1,7,#, and Heinz Jacobs2,# 1Division of Gene Regulation, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands 2Division of Tumor Biology & Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands 3Institute of Molecular Biology and Biotechnology, Bahauddin Zakariya University, 60800 Multan, Pakistan 4Division of Gene Regulation, Netherlands Cancer Institute, 1066CX Amsterdam, and Oncode Institute, The Netherlands 5Division of Tumor Biology & Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, and Oncode Institute, The Netherlands 6Department of Molecular Cell Biology and Immunology, Amsterdam UMC, Location VUmc, 1081HV Amsterdam, The Netherlands 7Department of Medical Biology, Amsterdam UMC, location AMC, UvA, 1105 AZ Amsterdam, The Netherlands * These authors contributed equally to this work. # Equal contribution and corresponding authors [email protected]; [email protected] Lead contact: Fred van Leeuwen 1 bioRxiv preprint doi: https://doi.org/10.1101/826255; this version posted November 18, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Human Transcription Factor Protein-Protein Interactions in Health and Disease
HELKA GÖÖS GÖÖS HELKA Recent Publications in this Series 45/2019 Mgbeahuruike Eunice Ego Evaluation of the Medicinal Uses and Antimicrobial Activity of Piper guineense (Schumach & Thonn) 46/2019 Suvi Koskinen AND DISEASE IN HEALTH INTERACTIONS PROTEIN-PROTEIN FACTOR HUMAN TRANSCRIPTION Near-Occlusive Atherosclerotic Carotid Artery Disease: Study with Computed Tomography Angiography 47/2019 Flavia Fontana DISSERTATIONES SCHOLAE DOCTORALIS AD SANITATEM INVESTIGANDAM Biohybrid Cloaked Nanovaccines for Cancer Immunotherapy UNIVERSITATIS HELSINKIENSIS 48/2019 Marie Mennesson Kainate Receptor Auxiliary Subunits Neto1 and Neto2 in Anxiety and Fear-Related Behaviors 49/2019 Zehua Liu Porous Silicon-Based On-Demand Nanohybrids for Biomedical Applications 50/2019 Veer Singh Marwah Strategies to Improve Standardization and Robustness of Toxicogenomics Data Analysis HELKA GÖÖS 51/2019 Iryna Hlushchenko Actin Regulation in Dendritic Spines: From Synaptic Plasticity to Animal Behavior and Human HUMAN TRANSCRIPTION FACTOR PROTEIN-PROTEIN Neurodevelopmental Disorders 52/2019 Heini Liimatta INTERACTIONS IN HEALTH AND DISEASE Efectiveness of Preventive Home Visits among Community-Dwelling Older People 53/2019 Helena Karppinen Older People´s Views Related to Their End of Life: Will-to-Live, Wellbeing and Functioning 54/2019 Jenni Laitila Elucidating Nebulin Expression and Function in Health and Disease 55/2019 Katarzyna Ciuba Regulation of Contractile Actin Structures in Non-Muscle Cells 56/2019 Sami Blom Spatial Characterisation of Prostate Cancer by Multiplex -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The E–Id Protein Axis Modulates the Activities of the PI3K–AKT–Mtorc1
Downloaded from genesdev.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press The E–Id protein axis modulates the activities of the PI3K–AKT–mTORC1– Hif1a and c-myc/p19Arf pathways to suppress innate variant TFH cell development, thymocyte expansion, and lymphomagenesis Masaki Miyazaki,1,8 Kazuko Miyazaki,1,8 Shuwen Chen,1 Vivek Chandra,1 Keisuke Wagatsuma,2 Yasutoshi Agata,2 Hans-Reimer Rodewald,3 Rintaro Saito,4 Aaron N. Chang,5 Nissi Varki,6 Hiroshi Kawamoto,7 and Cornelis Murre1 1Department of Molecular Biology, University of California at San Diego, La Jolla, California 92093, USA; 2Department of Biochemistry and Molecular Biology, Shiga University of Medical School, Shiga 520-2192, Japan; 3Division of Cellular Immunology, German Cancer Research Center, D-69120 Heidelberg, Germany; 4Department of Medicine, University of California at San Diego, La Jolla, California 92093, USA; 5Center for Computational Biology, Institute for Genomic Medicine, University of California at San Diego, La Jolla, California 92093, USA; 6Department of Pathology, University of California at San Diego, La Jolla, California 92093, USA; 7Department of Immunology, Institute for Frontier Medical Sciences, Kyoto University, Kyoto 606-8507, Japan It is now well established that the E and Id protein axis regulates multiple steps in lymphocyte development. However, it remains unknown how E and Id proteins mechanistically enforce and maintain the naı¨ve T-cell fate. Here we show that Id2 and Id3 suppressed the development and expansion of innate variant follicular helper T (TFH) cells. Innate variant TFH cells required major histocompatibility complex (MHC) class I-like signaling and were associated with germinal center B cells.