Formation and Structure of Radicals from D-Ribose and 2-Deoxy- D-Ribose by Reactions with SO 4 Radicals in Aqueous Solution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Reactivity of Fluorinated Radicals in Liquid Phase
THE REACnVITY OF FLUORINATED RADICALS IN LIQUID PHASE By XIAOXINRONG A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA . TO MOM, DAD AND XIAO JUN ACKNOWLEDGMENTS I wish to express my gratitude for the excellent guidance and friendship of my research advisor. Dr. William R. Dolbier, Jr. It is he who has made the fulfillment of this dream come true. His enthusiasm for chemistry and his understanding nature have been an inspiration to me throughout the course of my graduate studies. With Professor Dolbier, the chemistry is always surmountable and one comes away with a positive feeling towards oneself and the tasks ahead, which has led to an enjoyable and rewarding experience. I would like to give my special thanks to my committee members, especially. Dr. David H. Powell for giving me a chance to work in the mass-spectrum lab. in the early years of my graduate studies, which made me learn mass spectrometry systematically. Such knowledge has given me a lot of benefits in my research work. It is a pleasure to thank the many friends and colleagues I have met and worked with at the University of Florida. Due to my extended stay in the Dolbier group, thanks are required to a number of lab-mates; Conrad Burkholder for stimulating discussions, Michael Bartberger for his help in lab-management, Michelle Fletcher for her help in English to write this dissertation, Lu Lian, Rogelio Ocampo, Melvin Wagner, Luz Amalia, and more recently Xu Yuelian, Hao Jian, all for providing valuable friendships. -

A Dictionary of Chinese Characters: Accessed by Phonetics
A dictionary of Chinese characters ‘The whole thrust of the work is that it is more helpful to learners of Chinese characters to see them in terms of sound, than in visual terms. It is a radical, provocative and constructive idea.’ Dr Valerie Pellatt, University of Newcastle. By arranging frequently used characters under the phonetic element they have in common, rather than only under their radical, the Dictionary encourages the student to link characters according to their phonetic. The system of cross refer- encing then allows the student to find easily all the characters in the Dictionary which have the same phonetic element, thus helping to fix in the memory the link between a character and its sound and meaning. More controversially, the book aims to alleviate the confusion that similar looking characters can cause by printing them alongside each other. All characters are given in both their traditional and simplified forms. Appendix A clarifies the choice of characters listed while Appendix B provides a list of the radicals with detailed comments on usage. The Dictionary has a full pinyin and radical index. This innovative resource will be an excellent study-aid for students with a basic grasp of Chinese, whether they are studying with a teacher or learning on their own. Dr Stewart Paton was Head of the Department of Languages at Heriot-Watt University, Edinburgh, from 1976 to 1981. A dictionary of Chinese characters Accessed by phonetics Stewart Paton First published 2008 by Routledge 2 Park Square, Milton Park, Abingdon, OX14 4RN Simultaneously published in the USA and Canada by Routledge 270 Madison Ave, New York, NY 10016 Routledge is an imprint of the Taylor & Francis Group, an informa business This edition published in the Taylor & Francis e-Library, 2008. -

The Two Faces of the Guanyl Radical: Molecular Context and Behavior
molecules Review The Two Faces of the Guanyl Radical: Molecular Context and Behavior Chryssostomos Chatgilialoglu 1,2 1 ISOF, Consiglio Nazionale delle Ricerche, 40129 Bologna, Italy; [email protected]; Tel.: +39-051-6398309 2 Center of Advanced Technologies, Adam Mickiewicz University, 61-712 Pozna´n,Poland Abstract: The guanyl radical or neutral guanine radical G(-H)• results from the loss of a hydrogen atom (H•) or an electron/proton (e–/H+) couple from the guanine structures (G). The guanyl radical exists in two tautomeric forms. As the modes of formation of the two tautomers, their relationship and reactivity at the nucleoside level are subjects of intense research and are discussed in a holistic manner, including time-resolved spectroscopies, product studies, and relevant theoretical calculations. Particular attention is given to the one-electron oxidation of the GC pair and the complex mechanism of the deprotonation vs. hydration step of GC•+ pair. The role of the two G(-H)• tautomers in single- and double-stranded oligonucleotides and the G-quadruplex, the supramolecular arrangement that attracts interest for its biological consequences, are considered. The importance of biomarkers of guanine DNA damage is also addressed. Keywords: guanine; guanyl radical; tautomerism; guanine radical cation; oligonucleotides; DNA; G-quadruplex; time-resolved spectroscopies; reactive oxygen species (ROS); oxidation 1. The Guanine Sink Citation: Chatgilialoglu, C. The Two The free radical chemistry associated with guanine (Gua) and its derivatives, guano- Faces of the Guanyl Radical: sine (Guo), 2’-deoxyguanosine (dGuo), guanosine-50-monophosphate (GMP), and 20- Molecular Context and Behavior. deoxyguanosine-50-monophosphate (dGMP), is of particular interest due to its biological Molecules 2021, 26, 3511. -

Radical Hydroacylation of C-C and N-N Double Bonds in Air
University College London Radical Hydroacylation of C-C and N-N Double Bonds in Air by Jenna Marie Ahern Submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy Declaration I, Jenna Marie Ahern, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Jenna Marie Ahern October 2010 Radical Hydroacylation of C-C and N-N Double Bonds in Air Jenna Marie Ahern Abstract The formation of C-C and C-N bonds in modern organic synthesis is a key target for methodological advancement. Current methods of C-C and C-N bond formation often involve the use of expensive catalysts, or sub-stoichiometric reagents, which can lead to the generation of undesirable waste products. This thesis describes a novel and environmentally benign set of reaction conditions for the formation of C-C and C-N bonds by hydroacylation and this is promoted by mixing two reagents, an aldehyde and an electron-deficient double bond, under freely available atmospheric oxygen at room temperature Chapter 1 will provide an introduction to the thesis and mainly discusses methods for C-C bond formation, in particular, radical chemistry and hydroacylation. Chapter 2 describes the hydroacylation of vinyl sulfonates and vinyl sulfones (C-C double bonds) with aliphatic and aromatic aldehydes with a discussion and evidence for the mechanism of the transformation. Chapter 3 details the synthesis of precursors for intramolecular cyclisations and studies into aerobic intramolecular cyclisations. Chapter 4 describes the hydroacylation of vinyl phosphonates (C-C double bonds) and diazocarboxylates (N-N double bonds) with aliphatic and aromatic aldehydes bearing functional groups. -

Conjugated, Carbon-Centered Radicals
molecules Review Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals Takashi Kubo Department of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan; [email protected] Received: 26 January 2019; Accepted: 11 February 2019; Published: 13 February 2019 Abstract: Recently, long-lived, organic radical species have attracted much attention from chemists and material scientists because of their unique electronic properties derived from their magnetic spin and singly occupied molecular orbitals. Most stable and persistent organic radicals are heteroatom-centered radicals, whereas carbon-centered radicals are generally very reactive and therefore have had limited applications. Because the physical properties of carbon-centered radicals depend predominantly on the topology of the π-electron array, the development of new carbon-centered radicals is key to new basic molecular skeletons that promise novel and diverse applications of spin materials. This account summarizes our recent studies on the development of novel carbon-centered radicals, including phenalenyl, fluorenyl, and triarylmethyl radicals. Keywords: π-conjugated radicals; hydrocarbon radicals; persistent; anthryl; phenalenyl; fluorenyl 1. Introduction Organic radical species are generally recognized as highly reactive, intermediate species. However, recently, functional materials taking advantage of the feature of open-shell electronic structure have attracted much attention from chemists and material scientists; therefore, the development of novel, long-lived, organic, radical species becomes more important [1–7]. Nitronyl nitroxides, galvinoxyl, and DPPH are well known as stable, organic, radical species, which are commercially available chemicals. These stable radical species are “heteroatom-centered radicals”, in which unpaired electrons are mainly distributed on heteroatoms. -

New Blatter-Type Radicals from a Bench-Stable Carbene
ARTICLE Received 6 Aug 2016 | Accepted 28 Feb 2017 | Published 15 May 2017 DOI: 10.1038/ncomms15088 OPEN New Blatter-type radicals from a bench-stable carbene Jacob A. Grant1, Zhou Lu2, David E. Tucker1, Bryony M. Hockin1, Dmitry S. Yufit1, Mark A. Fox1, Ritu Kataky1, Victor Chechik2 & AnnMarie C. O’Donoghue1 Stable benzotriazinyl radicals (Blatter’s radicals) recently attracted considerable interest as building blocks for functional materials. The existing strategies to derivatize Blatter’s radicals are limited, however, and synthetic routes are complex. Here, we report that an inexpensive, commercially available, analytical reagent Nitron undergoes a previously unrecognized transformation in wet acetonitrile in the presence of air to yield a new Blatter-type radical with an amide group replacing a phenyl at the C(3)-position. This one-pot reaction of Nitron provides access to a range of previously inaccessible triazinyl radicals with excellent benchtop stabilities. Mechanistic investigation suggests that the reaction starts with a hydrolytic cleavage of the triazole ring followed by oxidative cyclization. Several derivatives of Nitron were prepared and converted into Blatter-type radicals to test the synthetic value of the new reaction. These results significantly expand the scope of using functionalized benzotriazinyls as stable radical building blocks. 1 Department of Chemistry, Durham University, South Road, Durham DH1 3LE, UK. 2 Department of Chemistry, University of York, Heslington, York YO10 5DD, UK. Correspondence and requests for materials should be addressed to V.C. (email: [email protected]) or to A.M.C.O’D. (email: [email protected]). NATURE COMMUNICATIONS | 8:15088 | DOI: 10.1038/ncomms15088 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/ncomms15088 1 latter’s radical 1 (Fig. -

"Radical Stability --- Thermochemical Aspects" In
Radical Stability—Thermochemical Aspects Johnny Hioe and Hendrik Zipse Department of Chemistry, LMU M¨unchen, M¨unchen, Germany 1 INTRODUCTION is quite challenging. Kinetic data, in contrast, are much more difficult to predict by theory, while the The terms “transient” and “persistent” are used determination of reaction rates can be approached frequently in the scientific literature to describe experimentally with a variety of direct or indirect the kinetic properties of open shell systems in methods, at least for sufficiently fast reactions homogeneous solution.1–5 The hydroxyl radical (see Radical Kinetics and Clocks). Theory and (HO•, 1), for example, is a transient species of experiment pair up nicely in this respect, as a central importance in atmospheric chemistry (see combination of these approaches is able to provide Atmospheric Radical Chemistry), as well as one a comprehensive picture of thermodynamic and of the most important reactive oxygen species kinetic data. (ROS) in aqueous solution, whereas the nitroxide 2,2,6,6-tetramethylpiperidine-1-oxyl, TEMPO (2)is a persistent radical stable enough to be bottled and 2 DEFINITIONS OF RADICAL STABILITY sold in bulk (Figure 1) (see Nitroxides in Synthetic Radical Chemistry). The thermodynamic stability of C-centered radicals However, despite their widespread use, these can be defined in various ways and several options terms are not too helpful for a quantitative approach are discussed in the following.6–10 One of the to radical chemistry as they do not reflect the most often used definitions is based on hydrogen influence of thermochemical driving force and transfer reactions as shown in Scheme 1 for reaction • intrinsic reaction barrier on the observed lifetime. -

Characterization of the Vinyloxy Radical by Ns Time
PCCP View Article Online PAPER View Journal | View Issue Fragmentation of a dioxolanyl radical via nonstatistical reaction dynamics: characterization Cite this: Phys. Chem. Chem. Phys., 2018, 20,19819 of the vinyloxy radical by ns time-resolved laser flash photolysis† a b b b Go¨tz Bucher, ‡* Mukul Lal, Anup Rana and Michael Schmittel * The photochemistry of two Barton esters, one derived from a dioxolane carboxylic acid and the other from pivalic acid, was investigated by product analysis and nanosecond laser flash photolysis (LFP). As expected, photolysis of the pivalate ester resulted in formation of the pyridine-2-thiyl and the t-butyl radical. Photolysis of the Barton ester of 2,2-dimethyl-1,3-dioxolane-4-carboxylic acid, on the other hand, revealed a complex multi-step fragmentation. In addition to the pyridine-2-thiyl and dioxolanyl radical, we gained evidence for the formation of the vinyloxy radical, CH2QCHO . The latter was identified in the Creative Commons Attribution 3.0 Unported Licence. LFP by its p-complexes with benzene and diphenylether, its rapid quenching by electron-rich arenes and tri-n-butyl tin hydride, and its oxidative power in presence of trifluoroacetic acid as demonstrated by the oxidation of ferrocene to ferrocenium. Formation of CH2QCHO can be rationalized via Received 24th May 2018, fragmentation of the dioxolanyl radical. As the calculated barriers are too high for the reaction sequence Accepted 14th July 2018 to occur on the LFP time scale, we investigated the fragmentation of the photoexcited Barton ester via DOI: 10.1039/c8cp03311k Born–Oppenheimer molecular dynamics simulations. In one trajectory, we could observe all reaction steps including ring opening of the dioxolanyl radical, suggesting that the excess energy gained in the ester rsc.li/pccp cleavage and decarboxylation may lead to fragmentation of the hot dioxolanyl radical. -

Free Radical and Ionic Reactions of (Benzoylmethyl)Mercurials Shekhar V
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1991 Free radical and ionic reactions of (Benzoylmethyl)mercurials Shekhar V. Kulkarni Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Kulkarni, Shekhar V., "Free radical and ionic reactions of (Benzoylmethyl)mercurials " (1991). Retrospective Theses and Dissertations. 10048. https://lib.dr.iastate.edu/rtd/10048 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfihn master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. -

Functionalised Oximes: Emergent Precursors for Carbon-, Nitrogen- and Oxygen-Centred Radicals
molecules Review Functionalised Oximes: Emergent Precursors for Carbon-, Nitrogen- and Oxygen-Centred Radicals John C. Walton Received: 11 December 2015 ; Accepted: 30 December 2015 ; Published: 7 January 2016 Academic Editor: Roman Dembinski EaStCHEM School of Chemistry, University of St. Andrews, St. Andrews, Fife KY16 9ST, UK; [email protected]; Tel.: +44-01334-463-864; Fax: +44-01334-463-808 Abstract: Oxime derivatives are easily made, are non-hazardous and have long shelf lives. They contain weak N–O bonds that undergo homolytic scission, on appropriate thermal or photochemical stimulus, to initially release a pair of N- and O-centred radicals. This article reviews the use of these precursors for studying the structures, reactions and kinetics of the released radicals. Two classes have been exploited for radical generation; one comprises carbonyl oximes, principally oxime esters and amides, and the second comprises oxime ethers. Both classes release an iminyl radical together with an equal amount of a second oxygen-centred radical. The O-centred radicals derived from carbonyl oximes decarboxylate giving access to a variety of carbon-centred and nitrogen-centred species. Methods developed for homolytically dissociating the oxime derivatives include UV irradiation, conventional thermal and microwave heating. Photoredox catalytic methods succeed well with specially functionalised oximes and this aspect is also reviewed. Attention is also drawn to the key contributions made by EPR spectroscopy, aided by DFT computations, in elucidating the structures and dynamics of the transient intermediates. Keywords: oxime esters; oxime ethers; free radicals; organic synthesis; photochemical reactions; EPR spectroscopy; photoredox catalysis; N-heterocycles 1. Introduction A huge variety of compounds containing the carbonyl functional group is available from natural and commercial sources. -
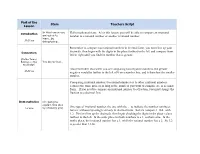
Part of the Lesson Stem Teachers Script Hello Mathematicians. After
Part of the Stem Teachers Script Lesson In this lesson you Introduction Hello mathematicians. After this lesson, you will be able to compare an irrational are going to number to a rational number or another irrational number. learn…by 10-20 sec doing/using… Remember to compare two rational numbers in decimal form, you must line up your decimals, then begin with the digits in the place furthest to the left and compare from Connection left to right until you find the number that is greater. (Define Terms/ Building on Prior You know that… Knowledge) Also remember, that when you are comparing two negative numbers, the greater 30-60 sec negative would be further to the left of 0 on a number line, and is therefore the smaller number. Comparing irrational numbers to rational numbers or to other irrational numbers requires the same process as long as the numbers you want to compare are in decimal form. If you need to compare an irrational number to a fraction, you must change the fraction to a decimal first. Demonstration I’m going to explain this idea One type of irrational number, the one with the ... to indicate the number continues 1-3 minc by showing you¦ forever without repeating is already in decimal form. So let's compare 1.124...with 1.2. First we line up the decimals, then begin checking the digits in the place values furthest to the left. In the units place for both numbers is a 1, so that's a tie. In the tenths place, the irrational number has a 1, while the rational number has a 2. -

Radicals and Radical Ions As Intermediates of Electron Transfer Processes Through Peptides
364 CHIMIA 2012, 66, No. 6 Organic Free radicals doi:10.2533/chimia.2012.364 Chimia 66 (2012) 364–367 © Schweizerische Chemische Gesellschaft Radicals and Radical Ions as Intermediates of Electron Transfer Processes through Peptides Bernd Giese*, Sonja Eckhardt, Miriam Lauz, Jian Gao, and Min Wang Abstract: Electron transfer (ET) through peptides and proteins is a key biochemical process, which involves radicals and radical ions as reactive intermediates. We have developed an assay that allows us to study this fundamental chemical reaction. Keywords: Electron transfer · Hopping · Peptide · Radical ion 1. Assay radical cation like 4 by chemically induced tween the electron acceptor and the elec- dynamic nuclear polarization (CIDNP).[6] tron donor.[4] Long-distance electron transfer (ET) Monte-Carlo simulations showed that the As shown by CD-spectroscopy the two through peptides, proteins and enzymes heterolytic cleavage 3➝4 is only possible triproline spacers induce the conforma- plays an important role in living organ- in the presence of polar solvents.[5b] The tion of a polyproline II (PPII) helix into isms.[1] More than a decade ago we studied last step of the reaction sequence is the the peptide, which does not change up to related reactions in DNA and concluded oxidation of the aromatic ring to radical 80 °C. The NMR spectra demonstrate that that long-distance ET through DNA can cation 5, which can be detected by UV/VIS 80% of the proline amide bonds are pres- occur only as a multistep hopping reac- spectroscopy (λ = 450 nm).[4] ent in trans-conformation, and molecular max tion.[2] To answer the question whether According to the mechanism shown in dynamic calculations predict the stretched an analogous hopping process also occurs Scheme 1, laser flash photolysis of 1 yields conformation 1b (Fig.