Renaissance of Sandmeyer-Type Reactions: Conversion of Aromatic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic Substitution
Dr. Tripti Gangwar AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic substitution ◦ The aromatic ring acts as a nucleophile, and attacks an added electrophile E+ ◦ An electron-deficient carbocation intermediate is formed (the rate- determining step) which is then deprotonated to restore aromaticity ◦ electron-donating groups on the aromatic ring (such as -OH, -OCH3, and alkyl) make the reaction faster, since they help to stabilize the electron-poor carbocation intermediate ◦ Lewis acids can make electrophiles even more electron-poor (reactive), increasing the reaction rate. For example FeBr3 / Br2 allows bromination to occur at a useful rate on benzene, whereas Br2 by itself is slow). In fact, a substitution reaction does occur! (But, as you may suspect, this isn’t an electrophilic aromatic substitution reaction.) In this substitution reaction the C-Cl bond breaks, and a C-O bond forms on the same carbon. The species that attacks the ring is a nucleophile, not an electrophile The aromatic ring is electron-poor (electrophilic), not electron rich (nucleophilic) The “leaving group” is chlorine, not H+ The position where the nucleophile attacks is determined by where the leaving group is, not by electronic and steric factors (i.e. no mix of ortho– and para- products as with electrophilic aromatic substitution). In short, the roles of the aromatic ring and attacking species are reversed! The attacking species (CH3O–) is the nucleophile, and the ring is the electrophile. Since the nucleophile is the attacking species, this type of reaction has come to be known as nucleophilic aromatic substitution. n nucleophilic aromatic substitution (NAS), all the trends you learned in electrophilic aromatic substitution operate, but in reverse. -

The Automated Flow Synthesis of Fluorine Containing Organic Compounds
The automated flow synthesis of fluorine containing organic compounds by CHANTAL SCHOLTZ Submitted in partial fulfilment of the requirements for the degree PHILOSOPHAE DOCTOR In the Faculty of Natural & Agricultural Sciences UNIVERSITY OF PRETORIA PRETORIA Supervisor: Dr D.L. Riley February 2019 DECLARATION I, Chantal Scholtz declare that the thesis/dissertation, which I hereby submit for the degree PhD Chemistry at the University of Pretoria, is my own work and has not previously been submitted by me for a degree at this or any other tertiary institution. Signature :.......................................... Date :..................................... ii ACKNOWLEDGEMENTS I would herewith sincerely like to show my gratitude to the following individuals for their help, guidance and assistance throughout the duration of this project: My supervisor, Doctor Darren Riley, for his knowledge and commitment. Thank you for being a fantastic supervisor and allowing me the opportunity to learn so many valuable skills. My husband, Clinton, for all your love, support and patience and for always being there for me. You are the best. My family, for all the encouragement and support you gave me as well as always believing in me. I will always appreciate what you have done for me. Mr Drikus van der Westhuizen and Mr Johan Postma for their assistance at the Pelchem laboratories with product isolation and characterisation. Dr Mamoalosi Selepe for NMR spectroscopy services, Jeanette Strydom for XRF services and Gerda Ehlers at the UP library for her invaluable assistance. All my friends and colleagues for the continuous moral support, numerous helpful discussions and necessary coffee breaks. My colleagues at Chemical Process Technologies for their ongoing support and motivation, especially Dr Hannes Malan and Prof. -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

Synthesis of Organobromines As a Tool for Their Characterisation and Environmental Occurrence Assessment
Synthesis of organobromines as a tool for their characterisation and environmental occurrence assessment Andreas Rydén Department of Materials and Environmental Chemistry Stockholm University Stockholm 2013 i Doctoral Thesis 2013 Department of Materials and Environmental Chemistry Stockholm University SE-106 91 Stockholm Sweden Abstract Polybrominated diphenyl ethers (PBDEs) have been intensively used as flame retardants (FRs) and have become ubiquitous environmental pollutants. PBDEs form hydroxylated PBDEs (OH-PBDEs) as metabolites. Further, some OH-PBDEs and methoxy-PBDEs (MeO-PBDEs) are natural products. These are all compounds of environmental and health concern and it is therefore important to confirm their identity and to assess their environmental levels and toxicities. Hence, it is vital to obtain authentic reference standards of individual PBDEs and OH/MeO-PBDEs. The thesis main aim was to develop synthesis methods of congener specific PBDEs, OH- and MeO-PBDEs. The second aim was to identify and quantify PBDEs, OH- and MeO-PBDEs in environmental samples. The third was to propose an abbreviation system for FRs. O-Arylation of brominated phenols, using either symmetrical or unsymmetrical brominated diphenyliodonium salts, was selected for synthesis of PBDEs and OH- /MeO-PBDEs. A total of 16 MeO-PBDEs, 11 OH-PBDEs, 1 diMeO-PBDE and 1 EtO-MeO-PBDE were synthesised. Three novel unsymmetrical diaryliodonium triflates were synthesised and used in synthesis. Optimisations were made to construct a reliable general method for congener specific PBDE synthesis, which was used in the synthesis of 8 representative PBDE congeners. The products were generally characterised by electron ionisation mass spectrometry (EIMS) and nuclear magnetic resonance (NMR) spectroscopy. -
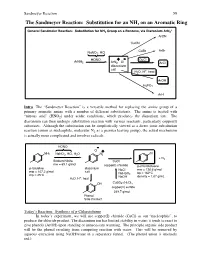
The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring
Sandmeyer Reaction 59 The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring + General Sandmeier Reaction: Substitution for NH2 Group on a Benzene, via Diazonium ArN2 ArCN CuCN CuBr ArBr NaNO2, HCl HONO + - CuCl ArNH2 ArN2 Cl ArCl diazonium salt + H2O, H , heat ArOH H3PO2 ArH Intro The “Sandmeyer Reaction” is a versatile method for replacing the amine group of a primary aromatic amine with a number of different substitutents. The amine is treated with “nitrous acid” (HNO2) under acidic conditions, which produces the diazonium ion. The diazonium can then undergo substitution reaction with various reactants, particularly copper(I) substrates. Although the substitution can be simplistically viewed as a direct ionic substitution reaction (anion as nucleophile, molecular N2 as a premier leaving group), the actual mechanism is actually more complicated and involves radicals. HONO - Cl + NH2 NaNO2, HCl, H2O N2 Cl + N2 Sodium Nitrite CuCl mw = 69.1 g/mol copper(I) chloride p-chlorotoluene p-toluidine diazonium NaCl mw = 126.6 g/mol mw = 107.2 g/mol salt NaHSO bp = 162ºC mp = 45ºC 3 + NaOH density = 1.07 g/mL H2O, H , heat OH CuSO4-(H2O)5 copper(II) sulfate 249.7 g/mol Phenol Side Product Today’s Reaction: Synthesis of p-Chlorotoluene In today’s experiment, we will use copper(I) chloride (CuCl) as our “nucleophile”, to produce the chloride product. The diazonium ion has limited stability in water; it tends to react to give phenols (ArOH) upon standing or unnecessary warming. The principle organic side product will be the phenol resulting from competing reaction with water. -

Sandmeyer Reaction
Sandmeyer reaction The Sandmeyer reaction is a chemical reaction used to synthesize aryl halides from Sandmeyer reaction aryl diazonium salts using copper salts as reagents or catalysts.[1][2][3] It is an example Named after Traugott of a radical-nucleophilic aromatic substitution. The Sandmeyer reaction provides a method through which one can perform unique transformations on benzene, such as Sandmeyer halogenation, cyanation, trifluoromethylation, and hydroxylation. Reaction type Substitution reaction Identifiers Organic sandmeyer- Chemistry reaction The reaction was discovered in 1884 by Swiss chemist Traugott Sandmeyer, when he Portal attempted to synthesize phenylacetylene from benzenediazonium chloride and cuprous RSC ontology RXNO:0000021 [4] acetylide. Instead, the main product he isolated was phenyl chloride. In modern ID times, the Sandmeyer reaction refers to any method for substitution of an aromatic amino group via preparation of its diazonium salt followed by its displacement with a nucleophile in the presence of catalytic copper(I) salts. (Due to the low cost of copper salts, a stoichiometric amount is often employed for better reactivity even when catalysis is possible.) The most commonly employed Sandmeyer reactions are the chlorination, bromination, cyanation, and hydroxylation reactions using CuCl, CuBr, CuCN, and Cu2O, respectively. More recently, trifluoromethylation of diazonium salts has been developed and is referred to as a 'Sandmeyer-type' reaction. Diazonium salts also react with boronates, iodide, thiols, water, -

Chem 232 Lecture 28.Pdf
CHEM 232 University of Illinois Organic Chemistry I at Chicago UIC Lecture 28 Organic Chemistry 1 Professor Duncan Wardrop April 22, 2010 1 Today’s Lecture Topics Covered: 1. Aryl Halides - Bonding, Physical Properties and Reactions 2. Nucleophilic Aromatic Substitution of Chlorobenzene 3. Nucleophilic Aromatic Substitution: Addition-Elimination 4. Floxcin - Application of Nucleophilic Aromatic Substitution 5. Nucleophilic Aromatic Substitution: Elimination-Addition 2 What’s the Difference Between Ar- and Ph-? Phenyl refers specifically to this: Aryl is a general term for all aromatic ring systems: C N N O 3 Chapter 23 Aryl Halides 4 23.1 Bonding in Aryl Halides 5 Aryl Halides X Aryl halides are halides in which the halogen is attached directly to an aromatic ring. Carbon-halogen bonds in aryl halides are shorter and stronger than carbon- halogen bonds in alkyl halides. 6 Dissociation Energies of Selected Compounds Bond Energy: kJ/mol (kcal/mol) X = H X = Cl CH3CH2X sp3 410 (98) 339 (81) 2 H2C CHX sp 452 (108) 368 (88) X sp2 469 (112) 406 (97) 7 Resonance Picture X X X H H C-X bonds in aryl halides have more double bond character than C-X bonds in alkyl halides 8 23.2 Sources of Aryl Halides 9 Preparation of Aryl Halides Halogenation of arenes (Section 12.5) H Br FeBr3 Br2 HBr electrophilic aromatic substitution 10 Preparation of Aryl Halides The Sandmeyer reaction (Section 22.17) N H H N N Cl Cl NaNO2 CuCl O HCl, H2O O heat O N+ N+ N+ O– O– O– Primary Aryl Diazonium Aryl Chloride Arylamine Salt diazotization-nucleophilic aromatic substitution 11 Preparation of Aryl Halides The Schiemann reaction (Section 22.17) N H H N BF4 N F 1. -

Preparation of Arylsulfonyl Chlorides by Chlorosulfonylation of in Situ Generated Diazonium Salts Using a Continuous flow Reactor†
View Online PAPER www.rsc.org/obc | Organic & Biomolecular Chemistry Preparation of arylsulfonyl chlorides by chlorosulfonylation of in situ generated diazonium salts using a continuous flow reactor† Laia Malet-Sanz,*a,b Julia Madrzak,b Steven V. Leya and Ian R. Baxendalea Received 19th July 2010, Accepted 27th August 2010 DOI: 10.1039/c0ob00450b A new flow procedure for the preparation of arylsulfonyl chlorides from aniline starting materials is described. The reaction conditions are mild, requiring no added acid and are amenable to continuous flow processing, in a safe, easily scalable and less labour intensive way than the corresponding batch method. Introduction was needed for the formation of the diazonium ion, during its addition to the SO2/AcOH/CuCl mixture and also throughout Sulfonyl chlorides are versatile precursors for the synthesis of the work-up process to ensure good yields.14 The reaction in 1 several important functional groups including sulfonamides, this format does raise some safety considerations due to the 2 3 4 4,5 sulfonyl fluorides, sulfonate esters, sulfones, and sulfinic acids. potentially highly explosive diazonium intermediate, a large Consequently, improved procedures for their synthesis are highly associated exotherm and the evolution of stoichiometric quantities desirable. Current methods for the generation of arylsulfonyl of nitrogen. Despite these issues the reaction has been successfully chlorides can be broadly divided into two classes. The first involves conducted at multi-kilogram scale.1a A rigorous safety assessment 6 the chlorination of aryl sulfonic acids or chlorination/oxidation led the authors to dilute the diazotisation step with large quantities 7 8 of disulfides and thiophenols The second route employs the of acetonitrile in order to fully solubilise the intermediate diazo- direct electrophilic aromatic substitution typically with chloro- nium salt. -
2,4-Bis(Fluoroalkyl)
2,4-Bis(fluoroalkyl)quinoline-3-carboxylates as Tools for the Development of Potential Agrochemical Ingredients Fallia Aribi, Armen Panossian, Jean-Pierre Vors, Sergiy Pazenok, Frédéric Leroux To cite this version: Fallia Aribi, Armen Panossian, Jean-Pierre Vors, Sergiy Pazenok, Frédéric Leroux. 2,4- Bis(fluoroalkyl)quinoline-3-carboxylates as Tools for the Development of Potential Agrochemical Ingre- dients. European Journal of Organic Chemistry, Wiley-VCH Verlag, 2018, Organofluorine Chemistry in Europe, 2018 (27-28), pp.3792-3802. 10.1002/ejoc.201800375. hal-02105501 HAL Id: hal-02105501 https://hal.archives-ouvertes.fr/hal-02105501 Submitted on 13 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 2,4-Bis(fluoroalkyl)quinoline-3-carboxylates as tools for the development of potential agrochemical ingredients Fallia Aribi,[a,b] Armen Panossian,[a,b] Jean-Pierre Vors,[b,c] Sergii Pazenok,[b,d] and Frédéric R. Leroux*[a,b] Abstract: From an easy and scalable synthetic access to quinoline groups appended to heteroaromatics may enhance the derivatives substituted by fluorinated groups in both C2- and C4- lipophilicity and oxidative stability of bioactive molecules.[5] positions developed in our laboratory, we devised the synthesis of a Despite this interest, only few series of quinolines substituted in new series of unprecedented 2,4-bis(fluoroalkyl)quinoline-3- C2- and C4-positions by fluorinated groups have been reported,[3c, carboxylates in two steps only. -

Application of the Sandmeyer Reaction Towards Synthesis of Selective
Anderson 1 Application of the Sandmeyer Reaction Towards Synthesis of Selective Toll-like Receptor 7 & 8 Antagonists Rachel J. Anderson Undergraduate Thesis March 27th, 2018 University of Colorado Boulder Thesis Advisor: Hang Hubert Yin, Ph.D.: Department of Chemistry & Biochemistry Committee Members: Xuedong Liu, Ph.D.: Department of Chemistry & Biochemistry Jeffrey Cameron, Ph.D.: Department of Chemistry & Biochemistry Karolin Luger, Ph.D.: Department of Chemistry & Biochemistry Pamela Harvey, Ph.D.: Molecular, Cellular & Developmental Biology Anderson 2 ACKNOWLEDGEMENTS I would like to begin by thanking Professor Hubert Yin and my lab mates in the Yin lab for putting up with my antics over the past two years and providing a great place to learn. This project would not have been possible without the extensive support and guidance of my direct mentor, Dr. Rosaura Padilla-Salinas, who has invested untold hours into my success. I would like to specifically thank Josh Kamps, Shafer Soars, and Dr. Adam Csakai for feedback, advice, and moral support. I would also like to thank my thesis committee: Xuedong Liu, Jeffrey Cameron, Karolin Luger, and Pamela Harvey for their patience and assistance. Finally, I am grateful for financial support provided by the Undergraduate Research Opportunities Program (UROP) at the University of Colorado, Boulder which made eating possible. Anderson 3 TABLE OF CONTENTS 1. Abstract ...................................................................................................................................... -

Organo-Fluorine Chemistry II
Organo-fluorine chemistry II Edited by David O'Hagan Generated on 04 October 2021, 05:56 Imprint Beilstein Journal of Organic Chemistry www.bjoc.org ISSN 1860-5397 Email: [email protected] The Beilstein Journal of Organic Chemistry is published by the Beilstein-Institut zur Förderung der Chemischen Wissenschaften. This thematic issue, published in the Beilstein Beilstein-Institut zur Förderung der Journal of Organic Chemistry, is copyright the Chemischen Wissenschaften Beilstein-Institut zur Förderung der Chemischen Trakehner Straße 7–9 Wissenschaften. The copyright of the individual 60487 Frankfurt am Main articles in this document is the property of their Germany respective authors, subject to a Creative www.beilstein-institut.de Commons Attribution (CC-BY) license. Organo-fluorine chemistry II David O'Hagan Editorial Open Access Address: Beilstein Journal of Organic Chemistry 2010, 6, No. 36. School of Chemistry, University of St Andrews, Fife, KY16 9ST, doi:10.3762/bjoc.6.36 United Kingdom Received: 12 April 2010 Email: Accepted: 12 April 2010 David O'Hagan - [email protected] Published: 20 April 2010 Guest Editor: D. O'Hagan © 2010 O'Hagan; licensee Beilstein-Institut. License and terms: see end of document. It is a pleasure to be able to introduce this second Thematic I am delighted that all of the authors have agreed to submit such Series on ‘organo-fluorine chemistry’ within the Beilstein high quality contributions to render this Thematic Series Journal of Organic Chemistry. The series now embeds the substantial and make it such a success. subject firmly as a special interest area of the journal. -

The Use of Molecular Orbital Calculations and Electrochemistry To
The use of molecular orbital calculations and electrochemistry to predict the reduction pathways of organochlorine compounds by Frederick Arthur Beland A thesis submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Chemistry Montana State University © Copyright by Frederick Arthur Beland (1974) Abstract: The CNDO/2 molecular orbital method was used to investigate the electrochemical reduction of organochlorine compounds of environmental interest. As the degree of chlorination increased in chlorinated benzenes and biphenyls, the LUMO-ς and LUMO-π both decreased in energy. The HOMO of the radical anions for each of these species was always a ς orbital. The location of highest electron density in the LUMO-ς for all of the chlorobenzenes, DDT, lindane and heptachlor predicted which chlorine was lost during electrochemical reduction. The electron density distribution in higher unoccupied ς orbitals of DDT and heptachlor predicted the order of carbon-chlorine bond scission in succeeding reductions. Electrochemical reduction pathways correctly predicted the observed anaerobic degradation pathways for DDT, DTE, lindane and hexachlorobenzene. 2,3,4,5,6-Pentachlorobiphenyl and decachlorobiphenyl were resistant to anaerobic reduction. The first electrochemical reduction product of decachlorobiphenyl did undergo anaerobic reduction. During anaerobic degradation heptachlor lost the allylic chlorine first as opposed to the "anti" methylene bridge chlorine observed electrochemically. These results indicate that if a compound has an E2d more cathodic than -1.75 V (vs. SCE) in a DMSO-TEABr solvent system, it will not reduce in an anaerobic environment. Compounds with an E2d more anodic than this value may be reduced in the environment.