The Automated Flow Synthesis of Fluorine Containing Organic Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Palladium-Catalyzed Arene Functionalization Via Single- Electron Reduction of Selectfluor
Palladium-Catalyzed Arene Functionalization via Single- Electron Reduction of Selectfluor The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Mazzotti, Anthony Renato. 2016. Palladium-Catalyzed Arene Functionalization via Single-Electron Reduction of Selectfluor. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:33840680 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Palladium-Catalyzed Arene Functionalization via Single-Electron Reduction of Selectfluor A dissertation presented by Anthony Renato Mazzotti to The Department of Chemistry and Chemical Biology In partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Chemistry Harvard University Cambridge, Massachusetts July 2016 © Anthony Renato Mazzotti All Rights Reserved. Dissertation Advisor: Professor Tobias Ritter Anthony Renato Mazzotti Palladium-Catalyzed Arene Functionalization via Single-Electron Reduction of Selectfluor Abstract Palladium-catalysis is commonly used in the functionalization of aromatic rings, ranging from prefunctionalized substrates such as aryl metals or aryl (pseudo)halides to the direct functionalization of aromatic C–H bonds. Palladium-catalyzed -

Historical Group
Historical Group NEWSLETTER and SUMMARY OF PAPERS No. 64 Summer 2013 Registered Charity No. 207890 COMMITTEE Chairman: Prof A T Dronsfield | Prof J Betteridge (Twickenham, 4, Harpole Close, Swanwick, Derbyshire, | Middlesex) DE55 1EW | Dr N G Coley (Open University) [e-mail [email protected]] | Dr C J Cooksey (Watford, Secretary: Prof. J. W. Nicholson | Hertfordshire) School of Sport, Health and Applied Science, | Prof E Homburg (University of St Mary's University College, Waldegrave | Maastricht) Road, Twickenham, Middlesex, TW1 4SX | Prof F James (Royal Institution) [e-mail: [email protected]] | Dr D Leaback (Biolink Technology) Membership Prof W P Griffith | Dr P J T Morris (Science Museum) Secretary: Department of Chemistry, Imperial College, | Mr P N Reed (Steensbridge, South Kensington, London, SW7 2AZ | Herefordshire) [e-mail [email protected]] | Dr V Quirke (Oxford Brookes Treasurer: Dr J A Hudson | University) Graythwaite, Loweswater, Cockermouth, | Prof. H. Rzepa (Imperial College) Cumbria, CA13 0SU | Dr. A Sella (University College) [e-mail [email protected]] Newsletter Dr A Simmons Editor Epsom Lodge, La Grande Route de St Jean, St John, Jersey, JE3 4FL [e-mail [email protected]] Newsletter Dr G P Moss Production: School of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London E1 4NS [e-mail [email protected]] http://www.chem.qmul.ac.uk/rschg/ http://www.rsc.org/membership/networking/interestgroups/historical/index.asp 1 RSC Historical Group Newsletter No. 64 Summer 2013 Contents From the Editor 2 Obituaries 3 Professor Colin Russell (1928-2013) Peter J.T. -

Anomalous Products in the Halogenation Reactions of Vinca Alkaloids 1.949
Send Orders for Reprints to [email protected] 2639 Current Organic Chemistry, 2016, 20, 2639-2646 RESEARCH ARTICLE ISSN: 1385-2728 eISSN: 1875-5348 Impact Factor: Anomalous Products in the Halogenation Reactions of Vinca Alkaloids 1.949 BENTHAM SCIENCE András Keglevich,1 László Hegeds,2 Lilla Péter,1 Judit Gyenese,1 Csaba Szántay, Jr.,3 Zsófia Dubrovay,3+ Miklós Dékány,3 Áron Szigetvári,3 Ana Martins,4++ József Molnár,4 Attila Hunyadi,5 Péter Keglevich1* and László Hazai1* 1Department of Organic Chemistry and Technology, University of Technology and Economics, Budapest, Hungary, H-1111 Budapest, Gellért tér 4. Hungary; 2MTA–BME Organic Chemical Technology Research Group, Hungarian Academy of Sciences, Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, Budafoki út 8, H-1111 Budapest, Hungary; 3Spectroscopic Research Division, Gedeon Richter Plc., H-1475 Budapest 10, P. O. Box 27, Hungary; 4Department of Medical Microbiology and Immunobiology, University of Szeged, 6720 Szeged, Dóm tér 10., Hungary; 5Institute of Pharmacognosy, University of Szeged, H-6720 Szeged, Eötvös u. 6., Hungary Abstract: Halogenation reactions of vindoline and 14,15-dihydrovindoline and its hydro- chloric salt were investigated and the anomalous reductions were discussed. Performing the hydrogenation in the presence of chlorine-containing solvent, e.g. dichloromethane, hydrogenolysation reaction of chlorine also took place. In this case unexpected chlorin- ated product could be observed. Performing the hydrogenation reaction only in the pres- ence of methanol, the expected reduced derivative was obtained. Upon bromination of vindoline with excess NBS, oxidation products with ring contraction and developing an oxygen bridge were isolated. The fluorination reactions of vinblastine using Selectfluor® and xenon difluoride as the fluorination reagents were unsuccessful because of the de- composition of the starting material. -

AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic Substitution
Dr. Tripti Gangwar AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic substitution ◦ The aromatic ring acts as a nucleophile, and attacks an added electrophile E+ ◦ An electron-deficient carbocation intermediate is formed (the rate- determining step) which is then deprotonated to restore aromaticity ◦ electron-donating groups on the aromatic ring (such as -OH, -OCH3, and alkyl) make the reaction faster, since they help to stabilize the electron-poor carbocation intermediate ◦ Lewis acids can make electrophiles even more electron-poor (reactive), increasing the reaction rate. For example FeBr3 / Br2 allows bromination to occur at a useful rate on benzene, whereas Br2 by itself is slow). In fact, a substitution reaction does occur! (But, as you may suspect, this isn’t an electrophilic aromatic substitution reaction.) In this substitution reaction the C-Cl bond breaks, and a C-O bond forms on the same carbon. The species that attacks the ring is a nucleophile, not an electrophile The aromatic ring is electron-poor (electrophilic), not electron rich (nucleophilic) The “leaving group” is chlorine, not H+ The position where the nucleophile attacks is determined by where the leaving group is, not by electronic and steric factors (i.e. no mix of ortho– and para- products as with electrophilic aromatic substitution). In short, the roles of the aromatic ring and attacking species are reversed! The attacking species (CH3O–) is the nucleophile, and the ring is the electrophile. Since the nucleophile is the attacking species, this type of reaction has come to be known as nucleophilic aromatic substitution. n nucleophilic aromatic substitution (NAS), all the trends you learned in electrophilic aromatic substitution operate, but in reverse. -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

Reduction of Aldehydes and Ketones to Their Corresponding Alcohols in 1- Butyl-3-Methylimidazolium Tetrafluoroborate
REDUCTION OF ALDEHYDES AND KETONES TO THEIR CORRESPONDING ALCOHOLS IN 1- BUTYL-3-METHYLIMIDAZOLIUM TETRAFLUOROBORATE Josiane Ayingeneye Dissertation submitted in fulfilment of the academic requirements for the degree of Master of Science in the School of Chemistry and Physics, University of KwaZulu-Natal, Durban. Supervisor: Prof Vincent O. Nyamori February 2018 Abstract The use and release of volatile organic solvents (VOS) to the atmosphere have become detrimental to both human health and his environment. In addition, the non-recyclability and the excessive consumption of these solvents in chemical industry influence their high costs. This has incited the growing need towards the design of new processes and benign solvents, helping in minimizing these raised issues. Ionic liquids (ILs) which are acknowledged as green and environmentally friendly solvents, are assigned as promising alternatives for replacing these VOS. The review of some ILs aspects such as structure, synthesis methods, physicochemical properties, solvent applications in reduction reactions, and their recyclability gave an impressive trend and a motivation towards our work. Synthesis of 1-butyl-3- methylimidazolium tetrafluoroborate ([BMIM][BF4]), as typical IL and investigation of its solvent efficiency, in the reduction of aldehydes and ketones to their corresponding alcohols, were the core objectives of this research. [BMIM][BF4] was synthesized via microwave (MW) and conventional methods. The characterization of the synthesized [BMIM][BF4] was done by means of Fourrier transform infrared (FTIR), nuclear magnetic resonance (1H- and 13C-NMR,) and liquid chromatograph- mass spectroscopy (LC-MS) techniques. Physicochemical properties of [BMIM][BF4] related to its solvent application, such as water content, density, viscosity and thermal stability, were explored. -

Synthesis of Organobromines As a Tool for Their Characterisation and Environmental Occurrence Assessment
Synthesis of organobromines as a tool for their characterisation and environmental occurrence assessment Andreas Rydén Department of Materials and Environmental Chemistry Stockholm University Stockholm 2013 i Doctoral Thesis 2013 Department of Materials and Environmental Chemistry Stockholm University SE-106 91 Stockholm Sweden Abstract Polybrominated diphenyl ethers (PBDEs) have been intensively used as flame retardants (FRs) and have become ubiquitous environmental pollutants. PBDEs form hydroxylated PBDEs (OH-PBDEs) as metabolites. Further, some OH-PBDEs and methoxy-PBDEs (MeO-PBDEs) are natural products. These are all compounds of environmental and health concern and it is therefore important to confirm their identity and to assess their environmental levels and toxicities. Hence, it is vital to obtain authentic reference standards of individual PBDEs and OH/MeO-PBDEs. The thesis main aim was to develop synthesis methods of congener specific PBDEs, OH- and MeO-PBDEs. The second aim was to identify and quantify PBDEs, OH- and MeO-PBDEs in environmental samples. The third was to propose an abbreviation system for FRs. O-Arylation of brominated phenols, using either symmetrical or unsymmetrical brominated diphenyliodonium salts, was selected for synthesis of PBDEs and OH- /MeO-PBDEs. A total of 16 MeO-PBDEs, 11 OH-PBDEs, 1 diMeO-PBDE and 1 EtO-MeO-PBDE were synthesised. Three novel unsymmetrical diaryliodonium triflates were synthesised and used in synthesis. Optimisations were made to construct a reliable general method for congener specific PBDE synthesis, which was used in the synthesis of 8 representative PBDE congeners. The products were generally characterised by electron ionisation mass spectrometry (EIMS) and nuclear magnetic resonance (NMR) spectroscopy. -
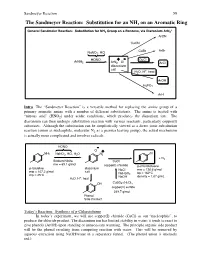
The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring
Sandmeyer Reaction 59 The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring + General Sandmeier Reaction: Substitution for NH2 Group on a Benzene, via Diazonium ArN2 ArCN CuCN CuBr ArBr NaNO2, HCl HONO + - CuCl ArNH2 ArN2 Cl ArCl diazonium salt + H2O, H , heat ArOH H3PO2 ArH Intro The “Sandmeyer Reaction” is a versatile method for replacing the amine group of a primary aromatic amine with a number of different substitutents. The amine is treated with “nitrous acid” (HNO2) under acidic conditions, which produces the diazonium ion. The diazonium can then undergo substitution reaction with various reactants, particularly copper(I) substrates. Although the substitution can be simplistically viewed as a direct ionic substitution reaction (anion as nucleophile, molecular N2 as a premier leaving group), the actual mechanism is actually more complicated and involves radicals. HONO - Cl + NH2 NaNO2, HCl, H2O N2 Cl + N2 Sodium Nitrite CuCl mw = 69.1 g/mol copper(I) chloride p-chlorotoluene p-toluidine diazonium NaCl mw = 126.6 g/mol mw = 107.2 g/mol salt NaHSO bp = 162ºC mp = 45ºC 3 + NaOH density = 1.07 g/mL H2O, H , heat OH CuSO4-(H2O)5 copper(II) sulfate 249.7 g/mol Phenol Side Product Today’s Reaction: Synthesis of p-Chlorotoluene In today’s experiment, we will use copper(I) chloride (CuCl) as our “nucleophile”, to produce the chloride product. The diazonium ion has limited stability in water; it tends to react to give phenols (ArOH) upon standing or unnecessary warming. The principle organic side product will be the phenol resulting from competing reaction with water. -

Durham E-Theses
Durham E-Theses Selective Fluorination Strategies BREEN, JESSICA,RUTH How to cite: BREEN, JESSICA,RUTH (2012) Selective Fluorination Strategies, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/3479/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk Abstract There is a great interest in the synthesis of fluorinated aromatic and heterocyclic compounds, which have a range of applications in the pharmaceutical industry. Many common routes to these compounds, however, are low yielding and or/expensive. This thesis is concerned with novel methods for the synthesis of fluoro-aromatics and fluoro- pyrazoles using conventional fluorinating agents, such as Selectfluor™, as well as using elemental fluorine and the flow reactor technology developed in Durham. Firstly, elemental fluorine was used to fluorinate a range of aromatics containing electron-donating substituents, using both batch and flow methods. -

Dissertation
DISSERTATION Titel Synthesis and Application of Fluorinated Carbohydrates and other Bioactive Compounds angestrebter akademischer Titel Doktorin der Naturwissenschaften (Dr. rer. nat.) Verfasserin: DI Michaela Braitsch Matrikelnummer: 9726189 Dissertationsgebiet Chemie Betreuer: Univ. Prof. Dr. Walther Schmid Wien im Oktober 2009 für Nicolas 7. 12. 1989 – 21. 2. 2008 Erfolgsrezept Ich will das Geheimnis verraten, dass mich zum Ziel geführt hat. Meine Stärke liegt einzig und allein in meiner Beharrlichkeit. LOUIS PASTEUR DANKSAGUNG Die vorliegende Arbeit entstand im Zeitraum von Feburar 2005 bis Oktober 2009 am Institut für Organische Chemie der Universität Wien. An dieser Stelle möchte ich mich bei allen, die zum Entstehen und Gelingen dieser Arbeit beigetragen haben, recht herzlich bedanken: Meinem Betreuer Prof. Walther Schmid für die interessante Themenstellung, sowie seine Hilfe und Unterstützung über die gesamte Zeit. Meinen Arbeitskollegen Michael Fischer, Stefan Hader, Ralph Hollaus, Christoph Lentsch, Roman Lichtenecker, Michael Nagl, Norberth Neuwirth, ChrisTina Nowikow, Christoph Schmölzer, Helga Wolf für die freundschaftliche Zusammenarbeit und das kollegiale Arbeitsklima. Den fleißigen Bienchen Gerlinde Benesch, Martina Drescher und Jale Özgur fürs Organisieren des Laborhaushaltes. Der NMR‐Abteilung Hanspeter Kählig, Lothar Brecker und Susanne Felsinger fürs Messen von zahlreichen Spektren. Der HPLC‐ und MS‐Abteilung Sabine Schneider und Peter Unteregger. Meiner Familie, im speziellen meinen Eltern und meiner Schwester Cornelia -

Download Date 07/10/2021 01:33:42
Functional ionic liquids in crystal engineering and drug delivery Item Type Thesis Authors Bansode, Ratnadeep V. Rights <a rel="license" href="http://creativecommons.org/licenses/ by-nc-nd/3.0/"><img alt="Creative Commons License" style="border-width:0" src="http://i.creativecommons.org/l/by- nc-nd/3.0/88x31.png" /></a><br />The University of Bradford theses are licenced under a <a rel="license" href="http:// creativecommons.org/licenses/by-nc-nd/3.0/">Creative Commons Licence</a>. Download date 07/10/2021 01:33:42 Link to Item http://hdl.handle.net/10454/14563 University of Bradford eThesis This thesis is hosted in Bradford Scholars – The University of Bradford Open Access repository. Visit the repository for full metadata or to contact the repository team © University of Bradford. This work is licenced for reuse under a Creative Commons Licence. FUNCTIONAL IONIC LIQUIDS FOR USE IN CRYSTAL ENGINEERING AND DRUG DELIVERY Ratnadeep Vitthal BANSODE Submitted for the Degree of Doctor of Philosophy School of Life Sciences University of Bradford 2016 Abstract Ratnadeep Vitthal Bansode Functional ionic liquids in crystal engineering and drug delivery Key words: Phosphonium ionic liquids, imidazolium ionic liquids, synthesis, pharmaceutical drugs, crystallisation, solubility, pharmaceuticals, API-ILs synthesis and drug release. The objective of this research is to explore the use of ionic liquds in crystal engineering and drug delivery. Ionic liquids have a wide range of applications in pharmaceutical field due to their unique physicochemical propertie ssuch as chemical, thermal stability, low melting point, nonvolatility, nonflamability, low toxicity and recyclability which offer unique and interesting potential for pharmaceuitcal applications. -

Sandmeyer Reaction
Sandmeyer reaction The Sandmeyer reaction is a chemical reaction used to synthesize aryl halides from Sandmeyer reaction aryl diazonium salts using copper salts as reagents or catalysts.[1][2][3] It is an example Named after Traugott of a radical-nucleophilic aromatic substitution. The Sandmeyer reaction provides a method through which one can perform unique transformations on benzene, such as Sandmeyer halogenation, cyanation, trifluoromethylation, and hydroxylation. Reaction type Substitution reaction Identifiers Organic sandmeyer- Chemistry reaction The reaction was discovered in 1884 by Swiss chemist Traugott Sandmeyer, when he Portal attempted to synthesize phenylacetylene from benzenediazonium chloride and cuprous RSC ontology RXNO:0000021 [4] acetylide. Instead, the main product he isolated was phenyl chloride. In modern ID times, the Sandmeyer reaction refers to any method for substitution of an aromatic amino group via preparation of its diazonium salt followed by its displacement with a nucleophile in the presence of catalytic copper(I) salts. (Due to the low cost of copper salts, a stoichiometric amount is often employed for better reactivity even when catalysis is possible.) The most commonly employed Sandmeyer reactions are the chlorination, bromination, cyanation, and hydroxylation reactions using CuCl, CuBr, CuCN, and Cu2O, respectively. More recently, trifluoromethylation of diazonium salts has been developed and is referred to as a 'Sandmeyer-type' reaction. Diazonium salts also react with boronates, iodide, thiols, water,