Organo-Fluorine Chemistry II
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
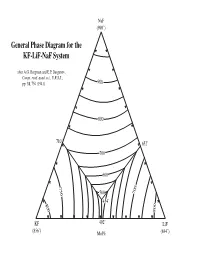
General Phase Diagram for the KF-Lif-Naf System
NaF (990˚) General Phase Diagram for the KF-LiF-NaF System after A.G. Bergman and E.P. Dergunov, Compt. rend. acad. sci., U.R.S.S., pp. 31, 754 (1941). 900 800 710˚ 652˚ 700 600 750 500 700 454˚ 800 800 KF 492˚ LiF (856˚) Mol% (844˚) Sodium Fluoride NaF 950˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 940˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 930˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 920˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 910˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 900˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 890˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 880˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 870˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 860˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 850˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 840˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 830˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 820˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 810˚ 810˚ ˚ KF 810 LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 800˚ 800˚ ˚ KF 800 LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 790˚ 790˚ ˚ KF 790 LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 780˚ 780˚ ˚ LiF KF 780 Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 770˚ 770˚ ˚ LiF KF 770 Potassium Fluoride -

Refrigerant Safety Refrigerant History
Refrigerant Safety The risks associated with the use of refrigerants in refrigeration and airconditioning equipment can include toxicity, flammability, asphyxiation, and physical hazards. Although refrigerants can pose one or more of these risks, system design, engineering controls, and other techniques mitigate this risk for the use of refrigerant in various types of equipment. Refrigerant History Nearly all of the historically used refrigerants were flammable, toxic, or both. Some were also highly reactive, resulting in accidents (e. g., leak, explosion) due to equipment failure, poor maintenance, or human error. The task of finding a nonflammable refrigerant with good stability was given to Thomas Midgley in 1926. With his associates Albert Leon Henne and Robert Reed McNary, Dr. Midgley observed that the refrigerants then in use comprised relatively few chemical elements, many of which were clustered in an intersecting row and column of the periodic table of elements. The element at the intersection was fluorine, known to be toxic by itself. Midgley and his collaborators felt, however, that compounds containing fluorine could be both nontoxic and nonflammable. The attention of Midgley and his associates was drawn to organic fluorides by an error in the literature that showed the boiling point for tetrafluoromethane (carbon tetrafluoride) to be high compared to those for other fluorinated compounds. The correct boiling temperature later was found to be much lower. Nevertheless, the incorrect value was in the range sought and led to evaluation of organic fluorides as candidates. The shorthand convention, later introduced to simplify identification of the organic fluorides for a systematic search, is used today as the numbering system for refrigerants. -

United States Patent Office Patented July 1, 1969
3,453,337 United States Patent Office Patented July 1, 1969 1. 2 3,453,337 FLUORINATION OF HALOGENATED The presence in the reaction mixture of the two fluo ORGANIC COMPOUNDS rides, or the complex fluoride enables better yields of Royston Henry Bennett and David Walter Cottrell, Ayon highly fluorinated products to be obtained under less mouth, England, assignors to Imperial Smelting Cor severe reaction condions, markedly increases the amount poration (N.S.C.) Limited, London, England, a British of fluorination reagent reacted under otherwise similar company conditions and enables the fluorination reaction to be No brawing. Filed Feb. 19, 1965, Ser. No. 434,128 carried out (for the same yield of product) at a lower Claims priority, application Great Britain, Feb. 26, 1964, temperature with the consequent use of less costly ma 7,932/64 terials and techniques of reactor construction. The pres Int, C. C07c 25/04 10 ence of the fluorides enables the vapor phase reaction U.S. C. 260-650 2 Claims to be carried out (for the same yields) at lower pressure This invention relates to the fluorination of organic than the pressure involved in the reactions using only halogen compounds and more especially to a process the alkali metal fluorides as proposed hitherto. The fur for the production of highly fluorinated aromatic com ther possibility of using a continuous flow apparatus such pounds by the replacement of higher halogen atoms in 15 as a fluidised reactor will be apparent to those familiar halogeno-aromatic compounds by fluorine atoms. with the art. The presence of the two fluorides or com Aromatic halogenocarbons containing carbon and halo plex fluoride enables a lower temperature to be em gen atoms only can be reacted with alkali fluorides ployed than was hitherto believed to be necessary, with under various conditions to give yields of halofluoro a consequent reduction in the extent of thermal degrada aromatic compounds. -

Development of Metal-Catalyzed Reactions of Allenes with Imines and the Investigation of Brθnsted Acid Catalyzed Ene Reactions
DEVELOPMENT OF METAL-CATALYZED REACTIONS OF ALLENES WITH IMINES AND THE INVESTIGATION OF BRΘNSTED ACID CATALYZED ENE REACTIONS BY LINDSEY O. DAVIS A Dissertation Submitted to the Graduate Faculty of WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY in the Department of Chemistry December 2009 Winston-Salem, North Carolina Approved By: Paul B. Jones, Ph.D., Advisor ____________________________ Examining Committee: Christa L. Colyer, Ph.D., chair ____________________________ S. Bruce King, Ph. D. _______________________________ Dilip K. Kondepudi, Ph.D. _______________________________ Suzanne L. Tobey, Ph.D. _______________________________ ACKNOWLEDGMENTS I must first acknowledge my family for their support throughout my education. My mother has always encouraged me to ask questions and seek answers, which has guided my inquisitive nature as a scientist. My grandparents have taught me the importance of character and my father taught me the value in hard work. I’d also like to thank my husband for moving away from Georgia, a sacrifice I greatly appreciate. Also, his cheerful disposition helped me stay relatively positive, especially when I had bad research days. My friends also played a very important role in keeping me positive during my time at Wake. Particularly my roommates put up with me more than most. I’d like to thank Lauren Eiter for her sense of humor, Tara Weaver for her taste in books and movies, and Jenna DuMond, for her encouragement and loyalty. Also I’ve made lifetime friends with Meredith and Kavita, and even though they left me at Wake, they were always willing to offer their support. -

2,4,6- Or 2,6-Alkoxyphenyl Dialkylphosphine, Tetrafluoroborate, Preparation Method and Use Thereof
(19) & (11) EP 2 492 274 A1 (12) EUROPEAN PATENT APPLICATION published in accordance with Art. 153(4) EPC (43) Date of publication: (51) Int Cl.: 29.08.2012 Bulletin 2012/35 C07F 9/50 (2006.01) B01J 31/24 (2006.01) C07C 43/215 (2006.01) C07C 41/30 (2006.01) (2006.01) (21) Application number: 09850497.0 C07D 307/58 (22) Date of filing: 21.12.2009 (86) International application number: PCT/CN2009/001527 (87) International publication number: WO 2011/047501 (28.04.2011 Gazette 2011/17) (84) Designated Contracting States: •LV,Bo AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Zhejiang 310027 (CN) HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL • FU, Chunling PT RO SE SI SK SM TR Zhejiang 310027 (CN) (30) Priority: 22.10.2009 CN 200910154029 (74) Representative: Meyer-Dulheuer, Karl-Hermann Dr. Meyer- Dulheuer & Partner (71) Applicant: Zhejiang University Patentanwaltskanzlei Hangzhou, Zhejiang 310027 (CN) Mainzer-Landstrasse 69-71 60329 Frankfurt am Main (DE) (72) Inventors: • MA, Shengming Zhejiang 310027 (CN) (54) 2,4,6- OR 2,6-ALKOXYPHENYL DIALKYLPHOSPHINE, TETRAFLUOROBORATE, PREPARATION METHOD AND USE THEREOF (57) The current invention relates to the structure, current invention uses only one step to synthesize dialkyl synthesis of dialkyl(2,4,6- or 2,6-alkoxyphenyl)phos- (2,4,6- or 2,6-alkoxyphenyl)phosphine and its tetrafluor- phine or its tetrafluoroborate, as well as its applications oborate is stable in the air. Compared with known syn- in the palladium catalyzed carbon-chlorine bond activa- thetic routes of ligands used in activating carbon- chlorine tion for Suzuki coupling reactions and carbon-nitrogen bonds, the method of current invention is short, easy to bond formation reactions. -

AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic Substitution
Dr. Tripti Gangwar AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic substitution ◦ The aromatic ring acts as a nucleophile, and attacks an added electrophile E+ ◦ An electron-deficient carbocation intermediate is formed (the rate- determining step) which is then deprotonated to restore aromaticity ◦ electron-donating groups on the aromatic ring (such as -OH, -OCH3, and alkyl) make the reaction faster, since they help to stabilize the electron-poor carbocation intermediate ◦ Lewis acids can make electrophiles even more electron-poor (reactive), increasing the reaction rate. For example FeBr3 / Br2 allows bromination to occur at a useful rate on benzene, whereas Br2 by itself is slow). In fact, a substitution reaction does occur! (But, as you may suspect, this isn’t an electrophilic aromatic substitution reaction.) In this substitution reaction the C-Cl bond breaks, and a C-O bond forms on the same carbon. The species that attacks the ring is a nucleophile, not an electrophile The aromatic ring is electron-poor (electrophilic), not electron rich (nucleophilic) The “leaving group” is chlorine, not H+ The position where the nucleophile attacks is determined by where the leaving group is, not by electronic and steric factors (i.e. no mix of ortho– and para- products as with electrophilic aromatic substitution). In short, the roles of the aromatic ring and attacking species are reversed! The attacking species (CH3O–) is the nucleophile, and the ring is the electrophile. Since the nucleophile is the attacking species, this type of reaction has come to be known as nucleophilic aromatic substitution. n nucleophilic aromatic substitution (NAS), all the trends you learned in electrophilic aromatic substitution operate, but in reverse. -

Download (4MB)
https://theses.gla.ac.uk/ Theses Digitisation: https://www.gla.ac.uk/myglasgow/research/enlighten/theses/digitisation/ This is a digitised version of the original print thesis. Copyright and moral rights for this work are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This work cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Enlighten: Theses https://theses.gla.ac.uk/ [email protected] SYNTHETIC AND BIOSYNTHETIC STUDIES ON SULPHUR-CONTAINING HETEROCYCLES A Thesis presented in part fulfilment of the requirement for the Degree of Doctor of Philosophy by Robert Andrew Lewis Department of Chemistry University of Glasgow September 1989 © ROBERT LEWIS 1989 ProQuest Number: 11003345 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11003345 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. -

The Automated Flow Synthesis of Fluorine Containing Organic Compounds
The automated flow synthesis of fluorine containing organic compounds by CHANTAL SCHOLTZ Submitted in partial fulfilment of the requirements for the degree PHILOSOPHAE DOCTOR In the Faculty of Natural & Agricultural Sciences UNIVERSITY OF PRETORIA PRETORIA Supervisor: Dr D.L. Riley February 2019 DECLARATION I, Chantal Scholtz declare that the thesis/dissertation, which I hereby submit for the degree PhD Chemistry at the University of Pretoria, is my own work and has not previously been submitted by me for a degree at this or any other tertiary institution. Signature :.......................................... Date :..................................... ii ACKNOWLEDGEMENTS I would herewith sincerely like to show my gratitude to the following individuals for their help, guidance and assistance throughout the duration of this project: My supervisor, Doctor Darren Riley, for his knowledge and commitment. Thank you for being a fantastic supervisor and allowing me the opportunity to learn so many valuable skills. My husband, Clinton, for all your love, support and patience and for always being there for me. You are the best. My family, for all the encouragement and support you gave me as well as always believing in me. I will always appreciate what you have done for me. Mr Drikus van der Westhuizen and Mr Johan Postma for their assistance at the Pelchem laboratories with product isolation and characterisation. Dr Mamoalosi Selepe for NMR spectroscopy services, Jeanette Strydom for XRF services and Gerda Ehlers at the UP library for her invaluable assistance. All my friends and colleagues for the continuous moral support, numerous helpful discussions and necessary coffee breaks. My colleagues at Chemical Process Technologies for their ongoing support and motivation, especially Dr Hannes Malan and Prof. -

123. Antimony
1998:11 The Nordic Expert Group for Criteria Documentation of Health Risks from Chemicals 123. Antimony John Erik Berg Knut Skyberg Nordic Council of Ministers arbete och hälsa vetenskaplig skriftserie ISBN 91–7045–471–x ISSN 0346–7821 http://www.niwl.se/ah/ah.htm National Institute for Working Life National Institute for Working Life The National Institute for Working Life is Sweden's center for research and development on labour market, working life and work environment. Diffusion of infor- mation, training and teaching, local development and international collaboration are other important issues for the Institute. The R&D competence will be found in the following areas: Labour market and labour legislation, work organization and production technology, psychosocial working conditions, occupational medicine, allergy, effects on the nervous system, ergonomics, work environment technology and musculoskeletal disorders, chemical hazards and toxicology. A total of about 470 people work at the Institute, around 370 with research and development. The Institute’s staff includes 32 professors and in total 122 persons with a postdoctoral degree. The National Institute for Working Life has a large international collaboration in R&D, including a number of projects within the EC Framework Programme for Research and Technology Development. ARBETE OCH HÄLSA Redaktör: Anders Kjellberg Redaktionskommitté: Anders Colmsjö och Ewa Wigaeus Hjelm © Arbetslivsinstitutet & författarna 1998 Arbetslivsinstitutet, 171 84 Solna, Sverige ISBN 91–7045–471–X ISSN 0346-7821 Tryckt hos CM Gruppen Preface The Nordic Council is an intergovernmental collaborative body for the five countries, Denmark, Finland, Iceland, Norway and Sweden. One of the committees, the Nordic Senior Executive Committee for Occupational Environmental Matters, initiated a project in order to produce criteria documents to be used by the regulatory authorities in the Nordic countries as a scientific basis for the setting of national occupational exposure limits. -

United States Patent Office Patented Sept
2,904,588 United States Patent Office Patented Sept. 15, 1959 - 2 m Three grams of the product prepared as described above was placed in a polyethylene bottle and 6 g. of water 2,904,588 was added. After the initial exothermic reaction had FLUOROPHOSPHORANES AND THEIR subsided and the solution had cooled, a white crystalline PREPARATION solid separated. This solid was recrystallized twice from water. After drying, the product melted at 159 to 161 William C. Smith, Wilmington, Del, assignor to E. I. du C. The melting point of benzenephosphonic acid is 159 Pont de Nemours and Company, Wilmington, Del., a to 161° C., and this is the product expected from the com corporation of Delaware plete hydrolysis of phenyltetrafluorophosphorane. No Drawing. Application March 26, 1956 O Nuclear magnetic resonance examination of the phenyl Serial No. 573,659 tetrafluorophosphorane showed it to have four equiv alent fluorine atoms bound to phosphorus, which indi 15 Claims. (C. 260-543) cated a square pyramidal structure. This invention relates to new compositions of matter 5 EXAMPLE II and to their preparation. Example I was repeated, using a charge consisting Organic fluorine compounds have attained considerable of 53.7 g (0.3 mole) of phenylphosphonous dichloride importance in recent years and simple and economic in the reaction flask and 65.1 g (0.3 mole) of antimony methods for their preparation are greatly desired. pentafluoride in the dropping funnel. The antimony This invention has as an object the preparation of new 20 pentafluoride was added to the phenylphosphonous di fluorophosphoranes. A further object is the provision of chloride at such a rate that the temperature of the reac a new process for their preparation. -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

Reaction of Potassium Fluoride with Organic Halogen Compounds. I
Reaction of Potassium Fluoride with Organic Halogen Compounds. I) Reactions of Potassium Fluoride with Organic Halides, Acids, aad Esters in presence ef Dimethyl Formamide and their Pyrolytic Decaboxylation in presence of Potassium Fluoride By You Sun Kim Atomic Energy Research Institute, Korea 有機 할로겐 化合物과 弗化加里의 反應 (第1報) 有機 할라어드, 酸 및 에스테르와 弗化加里의 디메칠 호쁨아마이드 溶蝶系 反應 및 高混■■脫炭酸-熱分解反應 金 裕 *善 (1963. 6. 19 受理) Abstract Reactions between potassium fluride with organic halogen-containning carboxylic acids in dimethyl formamide solvent gave a decarboxylation reaction for the case of fluoro carboxylic acids of the type of CF3 COOH, C3F7COOH, and C2F5COOH, whereas an additional partial fluorination together with dimeri zation reaction occured for the chlorine containning acids of the type of CH2CICOOH, CH3CHCICOOH, CHCI2COOH and o-Cl-CeHi-COOH. The phenyl halides showed no reactivity, but the halides with two electron attracting substituents on the benzene ring gave mainly dimerization reaction. The esters and alcohols gave an usual fluorination reaction. The same reactions in absence of the solvent at the elevated temperature increase the yield of the dimerized product and gave the cyclized product, fluorenone, in case of o-chlorobenzoic acid. It was found that the fluorination usually precede the decarboxylation reaction by checking the stiochemical sequence of reaction. Catalytic influence of potassium fluoride were discussed and the mechanism of the reaction was considered. 耍 約 「디메望호름아마이드」溶媒系에서 有機含할로겐化合物을 弗化加里와 反應시켜 본 結果 CFsCOOH, CsF’COOH, CzFQOOH 와 같은 含弗素有機酸에서는 脫炭酸反應이 일어나며, 含鹽素有機酸, CH2C1COOH. CH3CHC1COOH, CHC12- COOH 및 o-CK사LCOOH 은 一部 弗化反應이 일어 나고 雙合어imerization) 反應이 隨伴된다는 것을 究明하였다.