Exploiting Polypharmacology for Drug Target Deconvolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -

A Genome-Wide Association Study of Idiopathic Dilated Cardiomyopathy in African Americans
Journal of Personalized Medicine Article A Genome-Wide Association Study of Idiopathic Dilated Cardiomyopathy in African Americans Huichun Xu 1,* ID , Gerald W. Dorn II 2, Amol Shetty 3, Ankita Parihar 1, Tushar Dave 1, Shawn W. Robinson 4, Stephen S. Gottlieb 4 ID , Mark P. Donahue 5, Gordon F. Tomaselli 6, William E. Kraus 5,7 ID , Braxton D. Mitchell 1,8 and Stephen B. Liggett 9,* 1 Division of Endocrinology, Diabetes and Nutrition, Department of Medicine, University of Maryland School of Medicine, Baltimore, MD 21201, USA; [email protected] (A.P.); [email protected] (T.D.); [email protected] (B.D.M.) 2 Center for Pharmacogenomics, Department of Internal Medicine, Washington University School of Medicine, St. Louis, MO 63110, USA; [email protected] 3 Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, MD 21201, USA; [email protected] 4 Division of Cardiovascular Medicine, University of Maryland School of Medicine, Baltimore, MD 21201, USA; [email protected] (S.W.R.); [email protected] (S.S.G.) 5 Division of Cardiology, Department of Medicine, Duke University Medical Center, Durham, NC 27708, USA; [email protected] (M.P.D.); [email protected] (W.E.K.) 6 Department of Medicine, Division of Cardiology, Johns Hopkins University, Baltimore, MD 21218, USA; [email protected] 7 Duke Molecular Physiology Institute, Duke University Medical Center, Durham, NC 27701, USA 8 Geriatrics Research and Education Clinical Center, Baltimore Veterans Administration -

1 Kinobead Profiling Reveals Reprogramming of B-Cell Receptor Signaling in Response to Therapy Within Primary CLL Cells. Linley
bioRxiv preprint doi: https://doi.org/10.1101/841312; this version posted November 14, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Kinobead profiling reveals reprogramming of B-cell receptor signaling in response to therapy within primary CLL cells. Linley AJ1, Griffin R2, Cicconi S2, D’Avola A3$, MacEwan DJ4, Pettit AR1, Kalakonda N1, Packham G3, Prior IA5, Slupsky JR1. 1. Department of Molecular and Clinical Cancer Medicine, Institute of Translational Medicine, University of Liverpool, Sherrington Building, Ashton Street, Liverpool, UK. 2. CRUK Clinical Trials Unit, University of Liverpool, Waterhouse Building, Ashton Street, Liverpool. 3. Southampton Cancer Research UK Centre, Cancer Sciences Unit, Faculty of Medicine, University of Southampton, Southampton, UK. 4. Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, University of Liverpool, Sherrington Building, Ashton Street, Liverpool, UK. 5. Department of Cellular and Molecular Physiology, Institute of Translational Medicine, University of Liverpool, Nuffield Wing, Crown Street, Liverpool, UK. $ Current Address: The Francis Crick Institute, 1 Midland Road, London, UK. Corresponding author: Dr Adam J Linley, Department of Molecular and Clinical Cancer Medicine, Institute of Translational Medicine, University of Liverpool, Sherrington Building, Ashton Street, Liverpool, UK; [email protected]; +44(0)151 794 5310 Running head: Therapy brings about BCR signal changes. Key points 1. sIgM signaling patterns alter following in vivo therapy using either chemoimmunotherapy or ibrutinib. 2. Kinobeads provide a novel method for high-resolution investigation of signaling in primary CLL cells. 1 bioRxiv preprint doi: https://doi.org/10.1101/841312; this version posted November 14, 2019. -

Camk1d Kinase: a Potential Target for Breast Cancer Therapy by Jasmeen Kaur Sethi
CONFIDENTIAL CaMK1D kinase: a potential target for breast cancer therapy by Jasmeen Kaur Sethi A thesis submitted to the University of Birmingham for the degree of MRes in Cancer Sciences. School of Cancer Research College of Medical and Dental Sciences University of Birmingham August 2014 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. CONFIDENTIAL Abstract Basal like breast cancer (BLBC) is a highly aggressive and invasive subtype of triple negative breast cancer. BLBC patients are offered empirical treatments, which is associated with poor patient prognosis. The recent study of the genomic and transcriptomic background of 2000 breast cancers together with the discovery of a 10p13 chromosomal gain in BLBC suggest that a kinase known as CaMK1D serves as a driver for disease progression. In-house testing of small molecule compounds using structure-based techniques have already exhibited selective inhibition of the CaMK1D oncogenic state locking it in an autoinhibitory state. Here, we developed a cell-based model system for assays required to evaluate these inhibitors in vitro. BLBC cell lines, HCC70 and MDA-MB-231, were stably transformed using lentiviral transfections to express varying CaMK1D protein levels; knockdown, basal and overexpression. -

Emerging Applications of Metabolomic and Genomic Profiling in Diabetic Clinical Medicine
Reviews/Commentaries/ADA Statements REVIEW Emerging Applications of Metabolomic and Genomic Profiling in Diabetic Clinical Medicine AINE M. MCKILLOP, PHD however, powerful methodologies are now PETER R. FLATT, PHD available for exploitation of novel quantita- tive and qualitative disease-related biomark- ers. Novel biomarkers are needed that are Clinical and epidemiological metabolomics provides a unique opportunity to look at genotype- independent of known clinical risk factors. phenotype relationships as well as the body’s responses to environmental and lifestyle factors. fl Fundamentally, metabolomics aims to Fundamentally, it provides information on the universal outcome of in uencing factors on monitor changes in products of metabo- disease states and has great potential in the early diagnosis, therapy monitoring, and understand- lism and provide valuable information on ing of the pathogenesis of disease. Diseases, such as diabetes, with a complex set of interactions fl between genetic and environmental factors, produce changes in the body’s biochemical profile, arangeofin uencing factors and gene- thereby providing potential markers for diagnosis and initiation of therapies. There is clearly related outcomes. Exploitation of genomic a need to discover new ways to aid diagnosis and assessment of glycemic status to help reduce technology in recent times has resulted in diabetes complications and improve the quality of life. Many factors, including peptides, proteins, many technical advances, and genomic metabolites, nucleic acids, and polymorphisms, have been proposed as putative biomarkers for analysis has now emerged as a valuable diabetes. Metabolomics is an approach used to identify and assess metabolic characteristics, changes, tool in predicting the body’s response to and phenotypes in response to influencing factors, such as environment, diet, lifestyle, and path- fi stimuli caused by disease or injury. -

Rare Disruptive Variants in the DISC1 Interactome and Regulome: Association with Cognitive Ability and Schizophrenia
OPEN Molecular Psychiatry (2018) 23, 1270–1277 www.nature.com/mp ORIGINAL ARTICLE Rare disruptive variants in the DISC1 Interactome and Regulome: association with cognitive ability and schizophrenia S Teng1,2,10, PA Thomson3,4,10, S McCarthy1,10, M Kramer1, S Muller1, J Lihm1, S Morris3, DC Soares3, W Hennah5, S Harris3,4, LM Camargo6, V Malkov7, AM McIntosh8, JK Millar3, DH Blackwood8, KL Evans4, IJ Deary4,9, DJ Porteous3,4 and WR McCombie1 Schizophrenia (SCZ), bipolar disorder (BD) and recurrent major depressive disorder (rMDD) are common psychiatric illnesses. All have been associated with lower cognitive ability, and show evidence of genetic overlap and substantial evidence of pleiotropy with cognitive function and neuroticism. Disrupted in schizophrenia 1 (DISC1) protein directly interacts with a large set of proteins (DISC1 Interactome) that are involved in brain development and signaling. Modulation of DISC1 expression alters the expression of a circumscribed set of genes (DISC1 Regulome) that are also implicated in brain biology and disorder. Here we report targeted sequencing of 59 DISC1 Interactome genes and 154 Regulome genes in 654 psychiatric patients and 889 cognitively-phenotyped control subjects, on whom we previously reported evidence for trait association from complete sequencing of the DISC1 locus. Burden analyses of rare and singleton variants predicted to be damaging were performed for psychiatric disorders, cognitive variables and personality traits. The DISC1 Interactome and Regulome showed differential association across the phenotypes tested. After family-wise error correction across all traits (FWERacross), an increased burden of singleton disruptive variants in the Regulome was associated with SCZ (FWERacross P=0.0339). -

Discovery of Potent and Selective MRCK Inhibitors with Therapeutic
Published OnlineFirst January 30, 2018; DOI: 10.1158/0008-5472.CAN-17-2870 Cancer Translational Science Research Discovery of Potent and Selective MRCK Inhibitors with Therapeutic Effect on Skin Cancer Mathieu Unbekandt1, Simone Belshaw2, Justin Bower2, Maeve Clarke2, Jacqueline Cordes2, Diane Crighton2, Daniel R. Croft2, Martin J. Drysdale2, Mathew J. Garnett3, Kathryn Gill2, Christopher Gray2, David A. Greenhalgh4, James A.M. Hall3, Jennifer Konczal2, Sergio Lilla5, Duncan McArthur2, Patricia McConnell2, Laura McDonald2, Lynn McGarry6, Heather McKinnon2, Carol McMenemy4, Mokdad Mezna2, Nicolas A. Morrice5, June Munro1, Gregory Naylor1, Nicola Rath1, Alexander W. Schuttelkopf€ 2, Mairi Sime2, and Michael F. Olson1,7 Abstract The myotonic dystrophy–related Cdc42-binding kinases an autophosphorylation site, enabling development of a phos- MRCKa and MRCKb contribute to the regulation of actin–myosin phorylation-sensitive antibody tool to report on MRCKa status in cytoskeleton organization and dynamics, acting in concert with tumor specimens. In a two-stage chemical carcinogenesis model the Rho-associated coiled-coil kinases ROCK1 and ROCK2. The of murine squamous cell carcinoma, topical treatments reduced absence of highly potent and selective MRCK inhibitors has MRCKa S1003 autophosphorylation and skin papilloma out- resulted in relatively little knowledge of the potential roles of growth. In parallel work, we validated a phospho-selective anti- these kinases in cancer. Here, we report the discovery of the body with the capability to monitor drug pharmacodynamics. azaindole compounds BDP8900 and BDP9066 as potent and Taken together, our findings establish an important oncogenic selective MRCK inhibitors that reduce substrate phosphorylation, role for MRCK in cancer, and they offer an initial preclinical proof leading to morphologic changes in cancer cells along with inhi- of concept for MRCK inhibition as a valid therapeutic strategy. -

Systems Genetics Identifies a Role for Cacna2d1 Regulation in Elevated
Supplementary Dataset 1: Gene set enrichment analysis (GSEA) input Gene Symbol Gene name Score Cacna2d1 calcium channel, voltage-dependent, alpha 2/delta subunit 1 1 Pten phosphatase and tensin homolog (mutated in multiple advanced cancers 1) 0.8113829 Rora RAR-related orphan receptor A 0.7830256 Nfia nuclear factor I/A 0.77796155 Plxna2 plexin A2 0.7689365 Rwdd4a RWD domain containing 4A 0.75700486 Znf608 zinc finger protein 608 0.7334024 Huwe1 HECT, UBA and WWE domain containing 1 0.73015225 Prkg1 protein kinase, cGMP-dependent, type I 0.7254574 Rab39b RAB39B, member RAS oncogene family 0.71763426 St3gal2 ST3 beta-galactoside alpha-2,3-sialyltransferase 2 0.70345706 Lpgat1 lysophosphatidylglycerol acyltransferase 1 0.7029922 potassium voltage-gated channel, shaker-related subfamily, member 1 (episodic ataxia Kcna1 0.7010396 with myokymia) Ppp2ca protein phosphatase 2 (formerly 2A), catalytic subunit, alpha isoform 0.69497764 Maoa monoamine oxidase A 0.6936078 Socs6 suppressor of cytokine signaling 6 0.6922545 Abcc5 ATP-binding cassette, sub-family C (CFTR/MRP), member 5 0.6845195 Hspa4 heat shock 70kDa protein 4 0.6812371 Dctn4 dynactin 4 (p62) 0.68075943 Lats2 LATS, large tumor suppressor, homolog 2 (Drosophila) 0.67761165 Lin7a lin-7 homolog A (C. elegans) 0.673076 Itga9 integrin, alpha 9 0.6707624 Isoc1 isochorismatase domain containing 1 0.6701377 Cyfip2 cytoplasmic FMR1 interacting protein 2 0.6698394 Asip agouti signaling protein, nonagouti homolog (mouse) 0.6682124 Trpm7 transient receptor potential cation channel, subfamily M, -
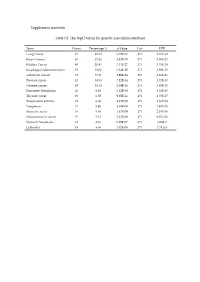
Supplement Materials Table S1. the Top15 Terms for Genetic Association
Supplement materials Table S1. The top15 terms for genetic association database. Term Count Percentage % p-Value List FDR Lung Cancer 73 25.34 6.70E-36 271 1.16E-32 Breast Cancer 67 23.26 2.92E-30 271 5.06E-27 Bladder Cancer 60 20.83 3.33E-27 271 5.78E-24 Esophageal adenocarcinoma 52 18.06 6.24E-29 271 1.08E-25 Colorectal Cancer 51 17.71 4.99E-24 271 8.66E-21 Prostate cancer 42 14.58 7.12E-14 271 1.23E-10 Ovarian cancer 39 13.54 2.04E-16 271 3.89E-13 Pancreatic Neoplasms 20 6.94 1.45E-08 271 2.52E-05 Thyroid cancer 19 6.59 9.98E-11 271 1.73E-07 Rheumatoid arthritis 19 6.60 8.82E-08 271 1.53E-04 Neoplasms 17 5.90 4.58E-08 271 7.94E-05 Stomach cancer 16 5.56 1.17E-09 271 2.03E-06 Head and neck cancer 15 5.21 5.23E-09 271 9.07E-06 Stomach Neoplasms 14 4.86 6.20E-07 271 1.08E-3 Leukemia 14 4.86 1.02E-06 271 1.78 E-3 Table S2. The results of the reverse docking for AutoDock Vina. PDB Protein Gene_Name Uniprot_ID Score 4TVJ Poly [ADP-ribose] polymerase 2 PARP2 Q9UGN5 -10.80 2DQ7 Proto-oncogene tyrosine-protein kinase Fyn FYN P06241 -10.00 4KIK Inhibitor of nuclear factor kappa-B kinase IKBKB O14920 -10.00 subunit beta 3EQR Activated CDC42 kinase 1 ACK1/TNK2 Q07912 -9.90 3MTF Activin receptor type-1 ACVR1 Q04771 -9.90 4GV0 Poly [ADP-ribose] polymerase 3 PARP3 Q9Y6F1 -9.90 1M6I Programmed cell death protein 8 AIF O95831 -9.80 1UA2 Cell division protein kinase 7 CDK7 P50613 -9.80 1Z6T Apoptotic protease activating factor 1 APAF1 O14727 -9.80 1ZXM DNA topoisomerase II TOPII P11388 -9.80 2WOU Serine/Threonine-protein kinase NEK7 NEK7 Q8TDX7 -9.60 -

Kinome Expression Profiling to Target New Therapeutic Avenues in Multiple Myeloma
Plasma Cell DIsorders SUPPLEMENTARY APPENDIX Kinome expression profiling to target new therapeutic avenues in multiple myeloma Hugues de Boussac, 1 Angélique Bruyer, 1 Michel Jourdan, 1 Anke Maes, 2 Nicolas Robert, 3 Claire Gourzones, 1 Laure Vincent, 4 Anja Seckinger, 5,6 Guillaume Cartron, 4,7,8 Dirk Hose, 5,6 Elke De Bruyne, 2 Alboukadel Kassambara, 1 Philippe Pasero 1 and Jérôme Moreaux 1,3,8 1IGH, CNRS, Université de Montpellier, Montpellier, France; 2Department of Hematology and Immunology, Myeloma Center Brussels, Vrije Universiteit Brussel, Brussels, Belgium; 3CHU Montpellier, Laboratory for Monitoring Innovative Therapies, Department of Biologi - cal Hematology, Montpellier, France; 4CHU Montpellier, Department of Clinical Hematology, Montpellier, France; 5Medizinische Klinik und Poliklinik V, Universitätsklinikum Heidelberg, Heidelberg, Germany; 6Nationales Centrum für Tumorerkrankungen, Heidelberg , Ger - many; 7Université de Montpellier, UMR CNRS 5235, Montpellier, France and 8 Université de Montpellier, UFR de Médecine, Montpel - lier, France ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2018.208306 Received: October 5, 2018. Accepted: July 5, 2019. Pre-published: July 9, 2019. Correspondence: JEROME MOREAUX - [email protected] Supplementary experiment procedures Kinome Index A list of 661 genes of kinases or kinases related have been extracted from literature9, and challenged in the HM cohort for OS prognostic values The prognostic value of each of the genes was computed using maximally selected rank test from R package MaxStat. After Benjamini Hochberg multiple testing correction a list of 104 significant prognostic genes has been extracted. This second list has then been challenged for similar prognosis value in the UAMS-TT2 validation cohort. -

Nanostring®: Product Data Sheet | Ncounter® GX Human Kinase
PRODUCT DATA SHEET nCounter® GX Human Kinase Kit nCounter® GX Human Kinase Kit Product Highlights Highly Curated • Our expert bio-informaticists use a very rigorous process in selecting the most meaningful set of genes Efficient • Multiplexed assay profiles 522 human kinase genes in a single reaction Cost-effective • Gold-standard data at a fraction of the cost Quick Turnaround Time • Complete kit with all consumables ready to ship next day nCounter® GX Human Kinase Kit The nCounter GX Human Kinase Kit is a comprehensive list of 522 human The nCounter Human Kinase Kit represents 99% of the KinBase content for genes known to be differentially expressed in the kinome. Human. With the nCounter GX Human Kinase Kit, scientists can leverage a pre-designed The final nCounter GX Human Kinase Kit consists of 522 protein kinase- panel to accelerate their research and quickly generate expression data for a related genes and 14 internal reference genes. For the gene list and additional large panel of protein kinase-related genes. information about this gene set, visit the nCounter Pre-built Panels product page at: www.nanostring.com. The gene list was compiled using the KinBase database at www.kinase.com. Home > Products > nCounter Gene Expression CodeSets > Pre-built Panels The database is based on the publication, The Protein Kinase Complement of the Human Genome, by G Manning, DB Whyte, R Martinez, T Hunter, S Sudarsanam (2002). Science 298:1912-1934. Molecules That Count® Translational Research Gene Expression miRNA Expression Copy Number Variation 1 PRODUCT DATA SHEET nCounter® GX Human Kinase Kit nCounter® Analysis System Overview The nCounter Analysis System from NanoString offers a cost-effective way to easily profile hundreds of gene transcripts simultaneously with high sensitivity and precision. -

Immuno-Detection by Sequencing Enables Large-Scale High-Dimensional Phenotyping in Cells
ARTICLE DOI: 10.1038/s41467-018-04761-0 OPEN Immuno-detection by sequencing enables large- scale high-dimensional phenotyping in cells Jessie A.G. van Buggenum 1, Jan P. Gerlach1, Sabine E.J. Tanis1, Mark Hogeweg1,2, Pascal W.T.C. Jansen3, Jesse Middelwijk4, Ruud van der Steen4, Michiel Vermeulen3, Hendrik G. Stunnenberg3, Cornelis A. Albers1,2 & Klaas W. Mulder1 Cell-based small molecule screening is an effective strategy leading to new medicines. Sci- 1234567890():,; entists in the pharmaceutical industry as well as in academia have made tremendous pro- gress in developing both large-scale and smaller-scale screening assays. However, an accessible and universal technology for measuring large numbers of molecular and cellular phenotypes in many samples in parallel is not available. Here we present the immuno- detection by sequencing (ID-seq) technology that combines antibody-based protein detec- tion and DNA-sequencing via DNA-tagged antibodies. We use ID-seq to simultaneously measure 70 (phospho-)proteins in primary human epidermal stem cells to screen the effects of ~300 kinase inhibitor probes to characterise the role of 225 kinases. The results show an association between decreased mTOR signalling and increased differentiation and uncover 13 kinases potentially regulating epidermal renewal through distinct mechanisms. Taken toge- ther, our work establishes ID-seq as a flexible solution for large-scale high-dimensional phenotyping in fixed cell populations. 1 Department of Molecular Developmental Biology, Radboud Institute for Molecular Life Sciences, Radboud University, PO Box 9101, 6500 HB Nijmegen, The Netherlands. 2 Department of Human Genetics, Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Center, PO Box 9101, 6500 HB Nijmegen, The Netherlands.