To Explain the General Mechanisms Leading to Glomerular Diseases And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Primer the Complement System
View metadata, citation and similar papers at core.ac.uk brought to you by CORE providedMagazine by ElsevierR259 - Publisher Connector vascular permeability and the infection. The second category Primer extravasation of plasma. This is in which there is persistent accompanied by the adhesion of formation of immune complexes is leukocytes to vascular endothelium autoimmune disease, in which, by The complement and their emigration into definition, the antigen is a system surrounding tissue. Complement permanent feature of the host. plays an important part in An understanding of how immune Philip Taylor, Marina inflammation, in helping to recruit complexes cause tissue injury effector cells and in promoting the developed from study of such Botto and Mark Walport killing and clearance of pathogens. diseases in humans and animals, and But complement can be both from the induction of experimental The system of plasma proteins called friend and foe. In some immune complex disease. In these complement was so-named because circumstances the activation of situations, immune complexes were it complements the activity of complement in response to immune shown to cause tissue injury through antibody in the lysis of bacteria. It complexes may cause harm: acutely activation of complement, as opsonin has a central role in host defence in the context of massive (binding to complement receptors on against many micro-organisms and in complement activation occurring in leukocytes) and as the source of the modulation of inflammatory patients with overwhelming Gram- anaphylatoxins (stimulating leukocyte reactions, an activity that has been negative bacterial sepsis, and chemotaxis and degranulation). But illuminated by the study of humans chronically, when an antibody the development of gene-targeted with naturally occurring deficiencies response develops in the context of mice with deficiencies of complement of complement. -

PATHOLOGY of the RENAL SYSTEM”, I Hope You Guys Like It
ﺑﺴﻢ اﷲ اﻟﺮﺣﻤﻦ اﻟﺮﺣﯿﻢ ھﺬه اﻟﻤﺬﻛﺮة ﻋﺒﺎرة ﻋﻦ إﻋﺎدة ﺗﻨﺴﯿﻖ وإﺿﺎﻓﺔ ﻧﻮﺗﺎت وﻣﻮاﺿﯿﻊ ﻟﻤﺬﻛﺮة زﻣﻼﺋﻨﺎ ﻣﻦ اﻟﺪﻓﻌﺔ اﻟﺴﺎﺑﻘﺔ ٤٢٧ اﻷﻋﺰاء.. ﻟﺘﺘﻮاﻓﻖ ﻣﻊ اﻟﻤﻨﮭﺞ اﻟﻤﻘﺮر ﻣﻦ اﻟﻘﺴﻢ ﺣﺮﺻﻨﺎ ﻓﯿﮭﺎ ﻋﻠﻰ إﻋﺎدة ﺻﯿﺎﻏﺔ ﻛﺜﯿﺮ ﻣﻦ اﻟﺠﻤﻞ ﻟﺘﻜﻮن ﺳﮭﻠﺔ اﻟﻔﮭﻢ وﺳﻠﺴﺔ إن ﺷﺎء اﷲ.. وﺿﻔﻨﺎ ﺑﻌﺾ اﻟﻨﻮﺗﺎت اﻟﻤﮭﻤﺔ وأﺿﻔﻨﺎ ﻣﻮاﺿﯿﻊ ﻣﻮﺟﻮدة ﺑﺎﻟـ curriculum ﺗﻌﺪﯾﻞ ٤٢٨ ﻋﻠﻰ اﻟﻤﺬﻛﺮة ﺑﻮاﺳﻄﺔ اﺧﻮاﻧﻜﻢ: ﻓﺎرس اﻟﻌﺒﺪي ﺑﻼل ﻣﺮوة ﻣﺤﻤﺪ اﻟﺼﻮﯾﺎن أﺣﻤﺪ اﻟﺴﯿﺪ ﺣﺴﻦ اﻟﻌﻨﺰي ﻧﺘﻤﻨﻰ ﻣﻨﮭﺎ اﻟﻔﺎﺋﺪة ﻗﺪر اﻟﻤﺴﺘﻄﺎع، وﻻ ﺗﻨﺴﻮﻧﺎ ﻣﻦ دﻋﻮاﺗﻜﻢ ! 2 After hours, or maybe days, of working hard, WE “THE PATHOLOGY TEAM” are proud to present “PATHOLOGY OF THE RENAL SYSTEM”, I hope you guys like it . Plz give us your prayers. Credits: 1st part = written by Assem “ THe AWesOme” KAlAnTAn revised by A.Z.K 2nd part = written by TMA revised by A.Z.K د.ﺧﺎﻟﺪ اﻟﻘﺮﻧﻲ 3rd part = written by Abo Malik revised by 4th part = written by A.Z.K revised by Assem “ THe AWesOme” KAlAnTAn 5th part = written by The Dude revised by TMA figures were provided by A.Z.K Page styling and figure embedding by: If u find any error, or u want to share any idea then plz, feel free to msg me [email protected] 3 Table of Contents Topic page THE NEPHROTIC SYNDROME 4 Minimal Change Disease 5 MEMBRANOUS GLOMERULONEPHRITIS 7 FOCAL SEGMENTAL GLOMERULOSCLEROSIS 9 MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS 11 DIABETIC NEPHROPATHY (new) 14 NEPHRITIC SYNDROME 18 Acute Post-infectious GN 19 IgA Nephropathy (Berger Disease) 20 Crescentic GN 22 Chronic GN 24 SLE Nephropathy (new) 26 Allograft rejection of the transplanted kidney (new) 27 Urinary Tract OBSTRUCTION, 28 RENAL STONES 23 HYDRONEPHROSIS -

Foundation Block Lecture Three Cell Mediated Immunity
Foundation Block Lecture Five Hypersensitivity Objectives: • To know that hypersensitivity reactions are over and excessive immune responses that can be harmful to body in four different ways • To be familiar with inflammatory processes in type I hypersensitivity reaction that mediates allergic inflammation • To recognize that type II hypersensitivity deals with immune responses against antigens that are integral part of cell membrane and are usually associated with autoimmune disorders • To know that type III hypersensitivity reactions are mediated by immune complexes and cause vasculitis • To describe type IV hypersensitivity is a purely cell mediated immune response associated with chronic inflammation ● Important. ● Extra notes. ● Females notes ● Males notes. What is hypersensitivity? Protective immunity: desirable reaction. Hypersensitivity: undesirable damaging reaction produced by excessive immune reactions. - Undesirable responses can be mediated by: - Antibody binding to antigens (Types I-III). - Cell mediated reaction to chemicals or proteins (Type IV). Types of hypersensitivity: 4 Types of hypersensitivity responses are classified by GEL AND COOMBS (names of scientists) according to the responding mechanisms, NOT the responding antigens: *From 433 team Type I: 1- Also termed as: - Immediate Hypersensitivity. - Allergic reactions. (e.g. asthma and eczema) - Anaphylactic reactions: severe and rapidly progressing systemic forms which can be quickly life threatening. …..............(causes.vasodilation and hypovolemia which causes heart stop then death) 2- Most people will not react to these allergens but some individuals “atopic” respond by producing large amounts of IgE in …........response to those otherwise harmless substances. 3- Non-allergic individuals respond to these allergens by producing IgG antibodies. 4- Features: - Antibody type: IgE - Cellular components: Mast cells, basophiles & eosinophils - Antigens: Also known as allergens ( antigens with low molecular weight & highly soluble). -

Suppression of Immune Complex-Induced Inflammation by the Chemotactic Factor Inactivator
Suppression of immune complex-induced inflammation by the chemotactic factor inactivator. K J Johnson, … , T P Anderson, P A Ward J Clin Invest. 1977;59(5):951-958. https://doi.org/10.1172/JCI108717. Research Article Small amounts (10(-10) mol) of purified human chemotactic factor inactivator (CFI) suppress leukocytic infiltration, permeability changes, and hemorrhage associated with acute immune complex-induced injury in rats. The reversed passive dermal Arthus reaction and acute immune complex-induced alveolitis in rats have served as the model systems of inflammation. The mechanism of inhibition does not appear to relate to interference with formation and deposition of immune complexes, or with fixation of complement in vitro or iv vivo. Human CFI inhibits in vitro the chemotactic activity generated in complement-activated rat serum. The inhibitory effects of human CFI are not seen if it is first heat inactivated. The data provide the first direct support for the conclusion that CFI has anti-inflammatory activity. Find the latest version: https://jci.me/108717/pdf Suppression of Immune Complex-Induced Inflammation by the Chemotactic Factor Inactivator KENT J. JOHNSON, THOMAS P. ANDERSON, and PETER A. WARD From the Department of Pathology, University of Connecticut Health Center, Farmington, Connecticut 06032 A B S T RA C T Small amounts (10-10 mol) of purified fested by skin tests (7-10). These observations have human chemotactic factor inactivator (CFI) suppress suggested that CFI is an important regulator of the leukocytic infiltration, permeability changes, and inflammatory response. In this communication direct hemorrhage associated with acute immune complex- evidence is presented to show that purified human CFI induced injury in rats. -

Glomerulonephritis Management in General Practice
Renal disease • THEME Glomerulonephritis Management in general practice Nicole M Isbel MBBS, FRACP, is Consultant Nephrologist, Princess Alexandra lomerular disease remains an important cause Hospital, Brisbane, BACKGROUND Glomerulonephritis (GN) is an G and Senior Lecturer in important cause of both acute and chronic kidney of renal impairment (and is the commonest cause Medicine, University disease, however the diagnosis can be difficult of end stage kidney disease [ESKD] in Australia).1 of Queensland. nikky_ due to the variability of presenting features. Early diagnosis is essential as intervention can make [email protected] a significant impact on improving patient outcomes. OBJECTIVE This article aims to develop However, presentation can be variable – from indolent a structured approach to the investigation of patients with markers of kidney disease, and and asymptomatic to explosive with rapid loss of kidney promote the recognition of patients who need function. Pathology may be localised to the kidney or further assessment. Consideration is given to the part of a systemic illness. Therefore diagnosis involves importance of general measures required in the a systematic approach using a combination of clinical care of patients with GN. features, directed laboratory and radiological testing, DISCUSSION Glomerulonephritis is not an and in many (but not all) cases, a kidney biopsy to everyday presentation, however recognition establish the histological diagnosis. Management of and appropriate management is important to glomerulonephritis (GN) involves specific therapies prevent loss of kidney function. Disease specific directed at the underlying, often immunological cause treatment of GN may require specialist care, of the disease and more general strategies aimed at however much of the management involves delaying progression of kidney impairment. -

Understanding the Immune System: How It Works
Understanding the Immune System How It Works U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES NATIONAL INSTITUTES OF HEALTH National Institute of Allergy and Infectious Diseases National Cancer Institute Understanding the Immune System How It Works U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES NATIONAL INSTITUTES OF HEALTH National Institute of Allergy and Infectious Diseases National Cancer Institute NIH Publication No. 03-5423 September 2003 www.niaid.nih.gov www.nci.nih.gov Contents 1 Introduction 2 Self and Nonself 3 The Structure of the Immune System 7 Immune Cells and Their Products 19 Mounting an Immune Response 24 Immunity: Natural and Acquired 28 Disorders of the Immune System 34 Immunology and Transplants 36 Immunity and Cancer 39 The Immune System and the Nervous System 40 Frontiers in Immunology 45 Summary 47 Glossary Introduction he immune system is a network of Tcells, tissues*, and organs that work together to defend the body against attacks by “foreign” invaders. These are primarily microbes (germs)—tiny, infection-causing Bacteria: organisms such as bacteria, viruses, streptococci parasites, and fungi. Because the human body provides an ideal environment for many microbes, they try to break in. It is the immune system’s job to keep them out or, failing that, to seek out and destroy them. Virus: When the immune system hits the wrong herpes virus target or is crippled, however, it can unleash a torrent of diseases, including allergy, arthritis, or AIDS. The immune system is amazingly complex. It can recognize and remember millions of Parasite: different enemies, and it can produce schistosome secretions and cells to match up with and wipe out each one of them. -

Immune Complex Tubulointerstitial Nephritis Due to Autoantibodies to the Proximal Tubule Brush Border
PATHOPHYSIOLOGY of the RENAL BIOPSY www.jasn.org Immune Complex Tubulointerstitial Nephritis Due to Autoantibodies to the Proximal Tubule Brush Border † ‡ Ivy A. Rosales,* A. Bernard Collins,* Paula Alves S. do Carmo, Nina Tolkoff-Rubin, R. Neal Smith,* and Robert B. Colvin* Department of *Pathology and ‡Division of Nephrology, Renal Transplantation Program, Massachusetts General Hospital, Boston, Massachusetts; and †Centro Especializado em Anatomia Patologica, Belo Horizonte, Minas Gerais, Brazil ABSTRACT Immune complex tubulointerstitial nephritis due to antibodies to brush border associated with focal tubulitis. Interstitial antigens of the proximal tubule has been demonstrated experimentally and rarely in fibrosis and tubular atrophy were diffuse humans. Our patient developed ESRD and early recurrence after transplantation. and present in 70% of the cortex. No gi- IgG and C3 deposits were conspicuous in the tubular basement membrane of antcellswereseen.Anarteryshowed proximal tubules, corresponding to deposits observed by electron microscopy. mild intimal fibrosis. Few IgG4-positive Rare subepithelial deposits were found in the glomeruli. The patient had no plasma cells were detected in the intersti- evidence of SLE and had normal complement levels. Serum samples from the tium (mean of 2 per high-power field). patient reacted with the brush border of normal human kidney, in contrast with the Immunofluorescence (IF) showed negative results with 20 control serum samples. Preliminary characterization of widespread granular deposits along the the brush border target antigen excluded megalin, CD10, and maltase. We postulate proximal tubular basement membrane that antibodies to brush border antigens cause direct epithelial injury, accumulate in (TBM), which stained for IgG (3+), C3 the tubular basement membrane, and elicit an interstitial inflammatory response. -
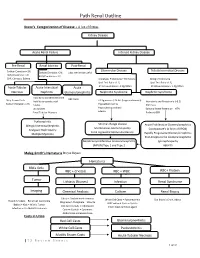
Path Renal Outline
Path Renal Outline Krane’s Categorization of Disease + A lot of Extras Kidney Disease Acute Renal Failure Intrinsic Kidney Disease Pre‐Renal Renal Intrinsic Post‐Renal Sodium Excretion <1% Glomerular Disease Tubulointerstitial Disease Sodium Excretion < 1% Sodium Excretion >2% Labs aren’t that useful BUN/Creatinine > 20 BUN/Creatinine < 10 CHF, Cirrhosis, Edema Urinalysis: Proteinuria + Hematuria Benign Proteinuria Spot Test Ratio >1.5, Spot Test Ratio <1.5, Acute Tubular Acute Interstitial Acute 24 Urine contains > 2.0g/24hrs 24 Urine contains < 1.0g/24hrs Necrosis Nephritis Glomerulonephritis Nephrotic Syndrome Nephritic Syndrome Inability to concentrate Urine RBC Casts Dirty Brown Casts Inability to secrete acid >3.5g protein / 24 hrs (huge proteinuria) Hematuria and Proteinuria (<3.5) Sodium Excretion >2% Edema Hypoalbuminemia RBC Casts Hypercholesterolemia Leukocytes Salt and Water Retention = HTN Focal Tubular Necrosis Edema Reduced GFR Pyelonephritis Minimal change disease Allergic Interstitial Nephritis Acute Proliferative Glomerulonephritis Membranous Glomerulopathy Analgesic Nephropathy Goodpasture’s (a form of RPGN) Focal segmental Glomerulosclerosis Rapidly Progressive Glomerulonephritis Multiple Myeloma Post‐Streptococcal Glomerulonephritis Membranoproliferative Glomerulonephritis IgA nephropathy (MPGN) Type 1 and Type 2 Alport’s Meleg‐Smith’s Hematuria Break Down Hematuria RBCs Only RBC + Crystals RBC + WBC RBC+ Protein Tumor Lithiasis (Stones) Infection Renal Syndrome Imaging Chemical Analysis Culture Renal Biopsy Calcium -

Nephrology Clinical Undergraduate Training
School of Medicine Nephrology clinical undergraduate training All care is taken to ensure that the information in this handbook is correct at the time of going to print. Handbook Version 5.0 Date of Origin: March 2019 Contents OBJECTIVES OF ATTACHMENT ................................................................................................... 3 TEACHING STRUCTURE: 3RD MEDICAL YEAR CLINICAL MEDICINE ATTACHMENTS .......................... 3 PROPOSED ATTACHMENT TIMETABLE: ................................................................................................ 5 READING LIST AND WEBSITES ............................................................................................................. 6 2 3rd Medical Year SPECIALTY: Nephrology - www.tcd.ie/medicine/thkc/education/ CONSULTANT: Prof G. Mellotte, Prof C Wall, Dr P Lavin, Dr B Griffin, Prof M Little HOSPITAL: Trinity Health Kidney Centre, Tallaght Hospital YEAR OF COURSE: 3 OBJECTIVES OF ATTACHMENT During this attachment, a student is expected to understand: - The clinical presentation of renal disease, e.g., proteinuria, hypertension, haematuria and uraemia. - Normal regulation of body water and sodium by the RAAS and ADH and how abnormalities give rise to changes in water and sodium homeostasis - Normal values of electrolytes in blood and urine and the clinical sequelae of a derangement in these. - A basic understanding of the following conditions: a. Acute kidney injury (pre-renal, post renal or intrinsic renal) b. Chronic kidney disease, focusing on diabetic nephropathy c. Glomerulonephritis: nephrotic / nephritic syndrome d. Myeloma and the kidney - The management of acute and chronic renal failure, including preparation for dialysis. - Impact of renal failure on drug handling. Be able to: - Take a full and appropriate current and past medical history. - Construct a synopsis or problem list based on the clinical assessment of a patient - Discuss the range of clinical investigations available and understand how they may be used to inform the differential diagnosis. -

16 the Kidney J
16 The Kidney J. Charles Jennette FPO FPO FPO FPO CONGENITAL ANOMALIES IgA Nephropathy (Berger Disease) Renal Agenesis Anti-Glomerular Basement Membrane Ectopic Kidney Glomerulonephritis Horseshoe Kidney ANCA Glomerulonephritis Renal Dysplasia VASCULAR DISEASES CONGENITAL POLYCYSTIC KIDNEY DISEASES Renal Vasculitis Autosomal Dominant Polycystic Kidney Disease Hypertensive Nephrosclerosis (Benign Nephrosclerosis) (ADPKD) Malignant Hypertensive Nephropathy Autosomal Recessive Polycystic Kidney Disease Renovascular Hypertension (ARPKD) Thrombotic Microangiopathy Nephronophthisis–Medullary Cystic Disease Cortical Necrosis ACQUIRED CYSTIC KIDNEY DISEASE DISEASES OF TUBULES AND INTERSTITIUM GLOMERULAR DISEASES Acute Tubular Necrosis (ATN) Nephrotic Syndrome Pyelonephritis Nephritic Syndrome Analgesic Nephropathy Glomerular Inflammation and Immune Mechanisms Drug-Induced (Hypersensitivity) Acute Tubulointerstitial Minimal-Change Glomerulopathy Nephritis Focal Segmental Glomerulosclerosis (FSGS) Light-Chain Cast Nephropathy Membranous Glomerulopathy Urate Nephropathy Diabetic Glomerulosclerosis RENAL STONES (NEPHROLITHIASIS AND Amyloidosis UROLITHIASIS) Hereditary Nephritis (Alport Syndrome) OBSTRUCTIVE UROPATHY AND HYDRONEPHROSIS Thin Glomerular Basement Membrane Nephropathy RENAL TRANSPLANTATION Acute Postinfectious Glomerulonephritis MALIGNANT TUMORS OF THE KIDNEY Type I Membranoproliferative Glomerulonephritis Wilms’ Tumor (Nephroblastoma) Type II Membranoproliferative Glomerulonephritis Renal Cell Carcinoma (RCC) (Dense Deposit Disease) -

LECTURE: 10 Title: IMMUNOLOGICAL MECHANISM in TISSUE DAMAGE TYPE-III IMMUNE-COMPLEX MEDIATED HYPERSENSITIVITY
LECTURE: 10 Title: IMMUNOLOGICAL MECHANISM IN TISSUE DAMAGE TYPE-III IMMUNE-COMPLEX MEDIATED HYPERSENSITIVITY LEARNING OBJECTIVES: The student should be able to: • Define type III hypersensitivity reactions. • Identify the role of the immune complex in type III reactions, and indicate which cause more damage small or large immune complexes, and locate the possible deposition of each complex. • Compare type III and type I reactions. • List some of the immune complex diseases: - Autoimmune diseases: ♦ Systemic lupus erythematosus (SLE). ♦ Rheumatoid arthritis (RA). ♦ Hyperthyroidism. - Infectious agents: ♦ Infectious mononucleosis. ♦ Meningitis. ♦ Viral hepatitis (Chronic stage). ♦ Poststreptococcal glomerulonephritis. ♦ Streptokinase (bacterial enzymes) in cardiac patients. - Drugs: ♦ Sulfonamides + penicillin. • Determine either type III reactions act locally or systematically. • Explain the mechanism of local immune complex deposition and give one example. • Explain the mechanism of generalized immune complex deposition, and give one example. • Explain the main target organ (s) for type III reactions. • Describe the mechanism of activation of type III reaction. • List a diagnostic method, and some examples of clinical features of type III such as: - Arthus reactions (Local immune complex-deposition). - Serum sickness (Systemic immune complex-deposition). LECTURE REFRENCE: 1. TEXTBOOK: ROITT, BROSTOFF, MALE IMMUNOLOGY. 6th edition. Chapter 23. pp. 357-367. 2. HANDOUT. Hypersensitivity – Type III Immune complexes are formed every time antibody meets antigen and are removed by the mononuclear phagocyte system following complement activation. Persistence of antigen from continued infection or in autoimmune disease can lead to immune-complex disease. Immune complexes can form both in the circulation, leading to systemic disease, and at local sites such as the lung. Complement helps to disrupt antigen-antibody bonds and keeps immune complexes soluble. -

Fcγ Receptors As Regulators of Immune Responses
REVIEWS Fcγ receptors as regulators of immune responses Falk Nimmerjahn* and Jeffrey V. Ravetch‡ Abstract | In addition to their role in binding antigen, antibodies can regulate immune responses through interacting with Fc receptors (FcRs). In recent years, significant progress has been made in understanding the mechanisms that regulate the activity of IgG antibodies in vivo. In this Review, we discuss recent studies addressing the multifaceted roles of FcRs for IgG (FcγRs) in the immune system and how this knowledge could be translated into novel therapeutic strategies to treat human autoimmune, infectious or malignant diseases. Regulatory T cells A productive immune response results from the effec- of antigenic peptides that are presented on the surface A T cell subset that is capable tive integration of positive and negative signals that of DCs to cytotoxic T cells, T helper cells, and regulatory of suppressing the activity have an impact on both innate and adaptive immune T cells. Thus, FcγRs are involved in regulating a multitude of other antigen-specific T cells cells. When positive signals dominate, cell activation of innate and adaptive immune responses, which makes including autoreactive T cells. Depletion of regulatory and pro-inflammatory responses ensue, resulting in the them attractive targets for the development of novel T cells results in the loss of elimination of pathogenic microorganisms and viruses. immunotherapeutic approaches (FIG. 1). peripheral tolerance and the In the absence of such productive stimulation, cell activa- In this Review, we summarize our current under- development of autoimmune tion is blocked and active anti-inflammatory responses standing of the role of different members of the FcγR disease.