PATHOLOGY of the RENAL SYSTEM”, I Hope You Guys Like It
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

RENAL BIOPSY and GLOMERULONEPHRITIS by J
Postgrad Med J: first published as 10.1136/pgmj.35.409.604 on 1 November 1959. Downloaded from 604 RENAL BIOPSY AND GLOMERULONEPHRITIS By J. H. Ross, M.D., M.R.C.P. Senior Medical Registrar, The Connaught Hospital, and Assistant, The Mledical Unit, The London Hospital Introduction sufficient to establish a diagnosis in a few condi- Safe methods of percutaneous renal biopsy were tions such as amyloid disease and diabetic first described by Perez Ara (1950) and Iversen glomerulosclerosis, but cortex with at least io and Brun (I95I) who used aspiration techniques. glomeruli is necessary for an accurate assessment Many different methods have since been described; of the renal structure; occasionally, up to 40 the simplest is that of Kark and Muehrcke (1954) glomeruli may be present in a single specimen. who use the Franklin modification of the Vim- The revelation of gross structural changes in Silverman needle. the kidneys of patients who can be recognized Brun and Raaschou (1958) have recently re- clinically as having chronic glomerulonephritis is viewed the majority of publications concerned of little value and does not contribute to manage- with renal biopsy and have concluded that the risk ment or prognosis. In contrast, biopsy specimensProtected by copyright. to the patients is slight. They mentioned three from patients in the early stages of glomerulo- fatalities which have been reported but considered nephritis, particularly those who have developed that, in each case, death was probably due to the the nephrotic syndrome insidiously without disease process rather than the investigation. haematuria or renal failure or who showy no Retroperitoneal haematomata have been recorded evidence of disease other than proteinuria, may in patients with hypertension or uraemia but are provide useful information. -

Glomerulonephritis Management in General Practice
Renal disease • THEME Glomerulonephritis Management in general practice Nicole M Isbel MBBS, FRACP, is Consultant Nephrologist, Princess Alexandra lomerular disease remains an important cause Hospital, Brisbane, BACKGROUND Glomerulonephritis (GN) is an G and Senior Lecturer in important cause of both acute and chronic kidney of renal impairment (and is the commonest cause Medicine, University disease, however the diagnosis can be difficult of end stage kidney disease [ESKD] in Australia).1 of Queensland. nikky_ due to the variability of presenting features. Early diagnosis is essential as intervention can make [email protected] a significant impact on improving patient outcomes. OBJECTIVE This article aims to develop However, presentation can be variable – from indolent a structured approach to the investigation of patients with markers of kidney disease, and and asymptomatic to explosive with rapid loss of kidney promote the recognition of patients who need function. Pathology may be localised to the kidney or further assessment. Consideration is given to the part of a systemic illness. Therefore diagnosis involves importance of general measures required in the a systematic approach using a combination of clinical care of patients with GN. features, directed laboratory and radiological testing, DISCUSSION Glomerulonephritis is not an and in many (but not all) cases, a kidney biopsy to everyday presentation, however recognition establish the histological diagnosis. Management of and appropriate management is important to glomerulonephritis (GN) involves specific therapies prevent loss of kidney function. Disease specific directed at the underlying, often immunological cause treatment of GN may require specialist care, of the disease and more general strategies aimed at however much of the management involves delaying progression of kidney impairment. -

Guidelines on Urolithiasis
Guidelines on Urolithiasis C. Türk (chair), T. Knoll (vice-chair), A. Petrik, K. Sarica, A. Skolarikos, M. Straub, C. Seitz © European Association of Urology 2014 TABLE OF CONTENTS PAGE 1. METHODOLOGY 7 1.1 Introduction 7 1.2 Data identification 7 1.3 Evidence sources 7 1.4 Level of evidence and grade of recommendation 7 1.5 Publication history 8 1.5.2 Potential conflict of interest statement 8 1.6 References 8 2. CLASSIFICATION OF STONES 9 2.1 Stone size 9 2.2 Stone location 9 2.3 X-ray characteristics 9 2.4 Aetiology of stone formation 9 2.5 Stone composition 9 2.6 Risk groups for stone formation 10 2.7 References 11 3. DIAGNOSIS 12 3.1 Diagnostic imaging 12 3.1.1 Evaluation of patients with acute flank pain 12 3.1.2 Evaluation of patients for whom further treatment of renal stones is planned 13 3.1.3 References 13 3.2 Diagnostics - metabolism-related 14 3.2.1 Basic laboratory analysis - non-emergency urolithiasis patients 15 3.2.2 Analysis of stone composition 15 3.3 References 16 4. TREATMENT OF PATIENTS WITH RENAL COLIC 16 4.1 Renal colic 16 4.1.1 Pain relief 16 4.1.2 Prevention of recurrent renal colic 16 4.1.3 Recommendations for analgesia during renal colic 17 4.1.4 References 17 4.2 Management of sepsis in obstructed kidney 18 4.2.1 Decompression 18 4.2.2 Further measures 18 4.2.3 References 18 5. STONE RELIEF 19 5.1 Observation of ureteral stones 19 5.1.1 Stone-passage rates 19 5.2 Observation of kidney stones 19 5.3 Medical expulsive therapy (MET) 20 5.3.1 Medical agents 20 5.3.2 Factors affecting success of medical expulsive -

Guidelines on Urolithiasis
GUIDELINES ON UROLITHIASIS (Text update April 2010) Ch. Türk (chairman), T. Knoll (vice-chairman), A. Petrik, K. Sarica, C. Seitz, M. Straub, O. Traxer Eur Urol 2001 Oct;40(4):362-71 Eur Urol 2007 Dec;52(6):1610-31 J Urol 2007 Dec;178(6):2418-34 Methodology This update is based on the 2008 version of the Urolithiasis guidelines produced by the former guidelines panel. Literature searches were carried out in Medline and Embase for ran- domised control trials and systematic reviews published between January 1st, 2008, and November 30, 2009, and sig- nificant differences in diagnosis and treatment or in evidence level were considered. Introduction Urinary stone disease continues to occupy an important place in everyday urological practice. The average lifetime risk of stone formation has been reported in the range of 5-10%. Recurrent stone formation is a common problem with all types of stones and therefore an important part of the medical care of patients with stone disease. Classification and Risk Factors Different categories of stone formers can be identified based Urolithiasis 251 on the chemical composition of the stone and the severity of the disease (Table 1). Special attention must be given to those patients who are at high risk of recurrent stone formation (Table 2). Table 1: Categories of stone formers Stone composition Category Non-calcium Infection stones: INF stones Magnesium ammonium phosphate Ammonium urate Uric acid UR Sodium urate Cystine CY Calcium First time stone former, without residual So stones stone or stone fragments. First time stone former, with residual Sres stone or stone fragments. -
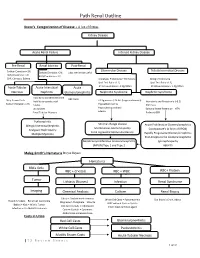
Path Renal Outline
Path Renal Outline Krane’s Categorization of Disease + A lot of Extras Kidney Disease Acute Renal Failure Intrinsic Kidney Disease Pre‐Renal Renal Intrinsic Post‐Renal Sodium Excretion <1% Glomerular Disease Tubulointerstitial Disease Sodium Excretion < 1% Sodium Excretion >2% Labs aren’t that useful BUN/Creatinine > 20 BUN/Creatinine < 10 CHF, Cirrhosis, Edema Urinalysis: Proteinuria + Hematuria Benign Proteinuria Spot Test Ratio >1.5, Spot Test Ratio <1.5, Acute Tubular Acute Interstitial Acute 24 Urine contains > 2.0g/24hrs 24 Urine contains < 1.0g/24hrs Necrosis Nephritis Glomerulonephritis Nephrotic Syndrome Nephritic Syndrome Inability to concentrate Urine RBC Casts Dirty Brown Casts Inability to secrete acid >3.5g protein / 24 hrs (huge proteinuria) Hematuria and Proteinuria (<3.5) Sodium Excretion >2% Edema Hypoalbuminemia RBC Casts Hypercholesterolemia Leukocytes Salt and Water Retention = HTN Focal Tubular Necrosis Edema Reduced GFR Pyelonephritis Minimal change disease Allergic Interstitial Nephritis Acute Proliferative Glomerulonephritis Membranous Glomerulopathy Analgesic Nephropathy Goodpasture’s (a form of RPGN) Focal segmental Glomerulosclerosis Rapidly Progressive Glomerulonephritis Multiple Myeloma Post‐Streptococcal Glomerulonephritis Membranoproliferative Glomerulonephritis IgA nephropathy (MPGN) Type 1 and Type 2 Alport’s Meleg‐Smith’s Hematuria Break Down Hematuria RBCs Only RBC + Crystals RBC + WBC RBC+ Protein Tumor Lithiasis (Stones) Infection Renal Syndrome Imaging Chemical Analysis Culture Renal Biopsy Calcium -

Factors in the Cause of Death in Carcinoma of the Cervix
FACTORS IN THE CAUSE OF DEATH IN CARCINOMA OF THE CERVIX A STUDY OF FIFTy-SEVEN CASES COMING TO NECROPSY B]ARNE PEARSON, M.D. (Prom the Cancer Institute and De;artment 01 Pathology, University of Minnesota Medkal School, Minneapolis ) This study is based upon 57 consecutive cases of carcinoma Qf the cervix in which autopsy examinations were performed. Thirty-one of these were studied at the University Hospitals. The remaining 26 were taken from the autopsy records in the Department of Pathology at the University of Minne sota. Microscopic sections were studied except in a few instances in which they could not be obtained. An attempt has been made to determine the im mediate lethal factors and the frequency with which they occur. No attempt is made to determine the efficacy of the treatment. URETERAL STRICTURE Stricture of the ureters with consequent hydronephrosis and hydroureters was the most striking and constant autopsy finding in this series of cases, being present in 42 cases, or 75 per cent. This is in accord with the observations of others. Ernst Holzbach in 4& carcinomas of the cervix coming to autopsy found 31 bilateral and 10 unilateral strictures. Sampson found that the fac tors responsible for strictures were external pressure of a mass of carcinoma around the ureters as a result of parametrial extension and lymphatic metasta sis, and added infection and edema. Carson states that metastasis may occur to the lymphatics of the ureters. Constriction by metastatic processes was not, however, very common in our series. It occurred only three times and was located in the midportion of the ureter. -

Nephrology Clinical Undergraduate Training
School of Medicine Nephrology clinical undergraduate training All care is taken to ensure that the information in this handbook is correct at the time of going to print. Handbook Version 5.0 Date of Origin: March 2019 Contents OBJECTIVES OF ATTACHMENT ................................................................................................... 3 TEACHING STRUCTURE: 3RD MEDICAL YEAR CLINICAL MEDICINE ATTACHMENTS .......................... 3 PROPOSED ATTACHMENT TIMETABLE: ................................................................................................ 5 READING LIST AND WEBSITES ............................................................................................................. 6 2 3rd Medical Year SPECIALTY: Nephrology - www.tcd.ie/medicine/thkc/education/ CONSULTANT: Prof G. Mellotte, Prof C Wall, Dr P Lavin, Dr B Griffin, Prof M Little HOSPITAL: Trinity Health Kidney Centre, Tallaght Hospital YEAR OF COURSE: 3 OBJECTIVES OF ATTACHMENT During this attachment, a student is expected to understand: - The clinical presentation of renal disease, e.g., proteinuria, hypertension, haematuria and uraemia. - Normal regulation of body water and sodium by the RAAS and ADH and how abnormalities give rise to changes in water and sodium homeostasis - Normal values of electrolytes in blood and urine and the clinical sequelae of a derangement in these. - A basic understanding of the following conditions: a. Acute kidney injury (pre-renal, post renal or intrinsic renal) b. Chronic kidney disease, focusing on diabetic nephropathy c. Glomerulonephritis: nephrotic / nephritic syndrome d. Myeloma and the kidney - The management of acute and chronic renal failure, including preparation for dialysis. - Impact of renal failure on drug handling. Be able to: - Take a full and appropriate current and past medical history. - Construct a synopsis or problem list based on the clinical assessment of a patient - Discuss the range of clinical investigations available and understand how they may be used to inform the differential diagnosis. -

16 the Kidney J
16 The Kidney J. Charles Jennette FPO FPO FPO FPO CONGENITAL ANOMALIES IgA Nephropathy (Berger Disease) Renal Agenesis Anti-Glomerular Basement Membrane Ectopic Kidney Glomerulonephritis Horseshoe Kidney ANCA Glomerulonephritis Renal Dysplasia VASCULAR DISEASES CONGENITAL POLYCYSTIC KIDNEY DISEASES Renal Vasculitis Autosomal Dominant Polycystic Kidney Disease Hypertensive Nephrosclerosis (Benign Nephrosclerosis) (ADPKD) Malignant Hypertensive Nephropathy Autosomal Recessive Polycystic Kidney Disease Renovascular Hypertension (ARPKD) Thrombotic Microangiopathy Nephronophthisis–Medullary Cystic Disease Cortical Necrosis ACQUIRED CYSTIC KIDNEY DISEASE DISEASES OF TUBULES AND INTERSTITIUM GLOMERULAR DISEASES Acute Tubular Necrosis (ATN) Nephrotic Syndrome Pyelonephritis Nephritic Syndrome Analgesic Nephropathy Glomerular Inflammation and Immune Mechanisms Drug-Induced (Hypersensitivity) Acute Tubulointerstitial Minimal-Change Glomerulopathy Nephritis Focal Segmental Glomerulosclerosis (FSGS) Light-Chain Cast Nephropathy Membranous Glomerulopathy Urate Nephropathy Diabetic Glomerulosclerosis RENAL STONES (NEPHROLITHIASIS AND Amyloidosis UROLITHIASIS) Hereditary Nephritis (Alport Syndrome) OBSTRUCTIVE UROPATHY AND HYDRONEPHROSIS Thin Glomerular Basement Membrane Nephropathy RENAL TRANSPLANTATION Acute Postinfectious Glomerulonephritis MALIGNANT TUMORS OF THE KIDNEY Type I Membranoproliferative Glomerulonephritis Wilms’ Tumor (Nephroblastoma) Type II Membranoproliferative Glomerulonephritis Renal Cell Carcinoma (RCC) (Dense Deposit Disease) -

Swiss Consensus Recommendations on Urinary Tract Infections in Children
European Journal of Pediatrics https://doi.org/10.1007/s00431-020-03714-4 REVIEW Swiss consensus recommendations on urinary tract infections in children Michael Buettcher1 & Johannes Trueck2 & Anita Niederer-Loher3 & Ulrich Heininger4 & Philipp Agyeman5 & Sandra Asner6 & Christoph Berger2 & Julia Bielicki4 & Christian Kahlert3 & Lisa Kottanattu7 & Patrick M. Meyer Sauteur2 & Paolo Paioni2 & Klara Posfay-Barbe8 & Christa Relly2 & Nicole Ritz4 & Petra Zimmermann9 & Franziska Zucol10 & Rita Gobet11 & Sandra Shavit12 & Christoph Rudin 13 & Guido Laube14 & Rodo von Vigier15 & Thomas J. Neuhaus16 Received: 28 July 2019 /Revised: 10 May 2020 /Accepted: 2 June 2020 # The Author(s) 2020, corrected publication 2020 Abstract The kidneys and the urinary tract are a common source of infection in children of all ages, especially infants and young children. The main risk factors for sequelae after urinary tract infections (UTI) are congenital anomalies of the kidney and urinary tract (CAKUT) and bladder-bowel dysfunction. UTI should be considered in every child with fever without a source. The differentiation between upper and lower UTI is crucial for appropriate management. Method of urine collection should be based on age and risk factors. The diagnosis of UTI requires urine analysis and significant growth of a pathogen in culture. Treatment of UTI should be based on practical considerations regarding age and presentation with adjustment of the initial antimicrobial treatment according to antimicrobial sensitivity testing. All children, regard- lessofage,shouldhaveanultrasoundoftheurinarytractperformed -

Silent Hydronephrosis/Pyonephrosis Due to Upper Urinary Tract Calculi in Spinal Cord Injury Patients
Spinal Cord (2000) 38, 661 ± 668 ã 2000 International Medical Society of Paraplegia All rights reserved 1362 ± 4393/00 $15.00 www.nature.com/sc Silent hydronephrosis/pyonephrosis due to upper urinary tract calculi in spinal cord injury patients S Vaidyanathan*,1, G Singh1, BM Soni1, P Hughes2, JWH Watt1, S Dundas3, P Sett1 and KF Parsons4 1Regional Spinal Injuries Centre, District General Hospital, Southport, Merseyside, UK; 2Department of Radiology, District General Hospital, Southport, Merseyside, UK; 3Department of Cellular Pathology, District General Hospital, Southport, Merseyside, UK; 4Department of Urology, Royal Liverpool University Hospital, Liverpool, UK Study design: A study of four patients with spinal cord injury (SCI) in whom a diagnosis of hydronephrosis or pyonephrosis was delayed since these patients did not manifest the traditional signs and symptoms. Objectives: To learn from these cases as to what steps should be taken to prevent any delay in the diagnosis and treatment of hydronephrosis/pyonephrosis in SCI patients. Setting: Regional Spinal Injuries Centre, Southport, UK. Methods: A retrospective review of cases of hydronephrosis or pyonephrosis due to renal/ ureteric calculus in SCI patients between 1994 and 1999, in whom there was a delay in diagnosis. Results: A T-5 paraplegic patient had two episodes of urinary tract infection (UTI) which were successfully treated with antibiotics. When he developed UTI again, an intravenous urography (IVU) was performed. The IVU revealed a non-visualised kidney and a renal pelvic calculus. In a T-6 paraplegic patient, the classical symptom of ¯ank pain was absent, and the symptoms of sweating and increased spasms were attributed to a syrinx. -

Jemds.Com Original Research Article
Jemds.com Original Research Article CLINICAL PROFILE AND SHORT-TERM OUTCOME OF ACUTE NEPHRITIC SYNDROME IN CHILDREN Surya Kandashamparambil Kamalakarababu1, Ansu Sam2, Sajini Varghese3 1Assistant Professor, Department of Paediatrics, Government Medical College, Kottayam. 2Senior Resident, Department of Paediatrics, Government Medical College, Kottayam. 3Assistant Professor, Department of Paediatrics, Government Medical College, Kottayam. ABSTRACT BACKGROUND Glomerulonephritis generally presents as a constellation of findings that includes haematuria, proteinuria and oedema. Poststreptococcal glomerulonephritis is the commonest form of acute glomerulonephritis in developing countries. Objectives- 1. To study the clinical profile of acute glomerulonephritis. 2. To study the different clinical laboratory parameters at admission and at 8 weeks of onset of illness. MATERIAL AND METHODS This is a prospective, descriptive study conducted in a tertiary care teaching hospital in South India from January 2016-December 2016. Data regarding the clinical features, laboratory parameters, treatment were collected. All these patients were further followed up at 8 weeks and descriptive analysis done. RESULTS The most common cause of glomerulonephritis in the study group was poststreptococcal glomerulonephritis (42.9%) followed by drug-induced nephropathy (28.6%). The most common complaints were haematuria, facial puffiness, decreased urine output and oedema. Hypertension was found only in 14.3% of study group. 4.8% of patients had nephrotic range of proteinuria. The most common infection preceding was Pyoderma (61.5%) in case of PSGN. High ASO titre was seen commonly with pharyngitis. 45.2% patients had decreased Serum C3 at diagnosis. All the patients were treated conservatively. At 8 weeks followup, haematuria was persisting in 28.6% patients. Only 2 patients had persistently low serum C3 and single patient had high creatinine. -

Since the Report by Maccalluim,L in I905, of Parathyroid Adenoma
ENLARGEMENT OF THE PARATHYROID GL.NDS IN RENAL DISEASE* A. M. PAPPENEIKER, M.D., ASiD S. L. WnyNs, M.D. (From Ike Department of Pathology, College of Physiciaxs axd Surgeons, Columbia Uxiversity, Newz York, N.Y.) The initial impetus to this study was given by a case of typical osteitis fibrosa with adenomatous enlargement of three parathyroid glands and associated polycystic kidneys.t The question which arose in the discussion was in regard to the relation between the kidney disease, obviously congenital in origin, and the parathyroid enlargement. Since the report by MacCalluim,l in I905, of parathyroid adenoma associated with chronic glomerulonephritis, the simultaneous occur- rence of renal lesions with parathyroid tumors or enlargement has been noted repeatedly. This association has been emphasized par- ticularly in the recent study of Albright, Baird, Cope and Bloom- berg,2 who collected 83 cases of hyperparathyroidism, 43 of which showed some type of renal damage. The renal lesions are attributed to the precpitation of calcium in the tubules, with resultant sclero- sis, contraction and insufficiency, or to the formation of calculi in the pelvis associated with pyelonephritis. Thus, these authors in general regard the renal lesions as a sequel to the chronic hyperparathyroidism and stress especially the de- position of calcium in the renal tissue as the proximate cause of the kidney lesions. However, in their discussion they raise the question as to whether the parathyroid enlargement may not be secondary to the renal disease in the cases in which multiple glands are affected. They report: "It seems conceivable that a chronic renal insuf- ficiency with phosphate retention and a high inorganic phosphorus level might likewise cause hyperplasia of all parathyroid tissue which might go on to multiple tumor formation...