Legionella Pneumophila Effector Function Using Proteomic Approaches
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NCTC) Bacterial Strain Equivalents to American Type Culture Collection (ATCC) Bacterial Strains
This list shows National Collection of Type Cultures (NCTC) bacterial strain equivalents to American Type Culture Collection (ATCC) bacterial strains. NCTC Number CurrentName ATCC Number NCTC 7212 Acetobacter pasteurianus ATCC 23761 NCTC 10138 Acholeplasma axanthum ATCC 25176 NCTC 10171 Acholeplasma equifetale ATCC 29724 NCTC 10128 Acholeplasma granularum ATCC 19168 NCTC 10172 Acholeplasma hippikon ATCC 29725 NCTC 10116 Acholeplasma laidlawii ATCC 23206 NCTC 10134 Acholeplasma modicum ATCC 29102 NCTC 10188 Acholeplasma morum ATCC 33211 NCTC 10150 Acholeplasma oculi ATCC 27350 NCTC 10198 Acholeplasma parvum ATCC 29892 NCTC 8582 Achromobacter denitrificans ATCC 15173 NCTC 10309 Achromobacter metalcaligenes ATCC 17910 NCTC 10807 Achromobacter xylosoxidans subsp. xylosoxidans ATCC 27061 NCTC 10808 Achromobacter xylosoxidans subsp. xylosoxidans ATCC 17062 NCTC 10809 Achromobacter xylosoxidans subsp. xylosoxidans ATCC 27063 NCTC 12156 Acinetobacter baumannii ATCC 19606 NCTC 10303 Acinetobacter baumannii ATCC 17904 NCTC 7844 Acinetobacter calcoaceticus ATCC 15308 NCTC 12983 Acinetobacter calcoaceticus ATCC 23055 NCTC 8102 acinetobacter dna group 13 ATCC 17903 NCTC 10304 Acinetobacter genospecies 13 ATCC 17905 NCTC 10306 Acinetobacter haemolyticus ATCC 17907 NCTC 10305 Acinetobacter haemolyticus subsp haemolyticus ATCC 17906 NCTC 10308 Acinetobacter johnsonii ATCC 17909 NCTC 10307 Acinetobacter junii ATCC 17908 NCTC 5866 Acinetobacter lwoffii ATCC 15309 NCTC 12870 Actinobacillus delphinicola ATCC 700179 NCTC 8529 Actinobacillus equuli ATCC 19392 -

Legionella Shows a Diverse Secondary Metabolism Dependent on a Broad Spectrum Sfp-Type Phosphopantetheinyl Transferase
Legionella shows a diverse secondary metabolism dependent on a broad spectrum Sfp-type phosphopantetheinyl transferase Nicholas J. Tobias1, Tilman Ahrendt1, Ursula Schell2, Melissa Miltenberger1, Hubert Hilbi2,3 and Helge B. Bode1,4 1 Fachbereich Biowissenschaften, Merck Stiftungsprofessur fu¨r Molekulare Biotechnologie, Goethe Universita¨t, Frankfurt am Main, Germany 2 Max von Pettenkofer Institute, Ludwig-Maximilians-Universita¨tMu¨nchen, Munich, Germany 3 Institute of Medical Microbiology, University of Zu¨rich, Zu¨rich, Switzerland 4 Buchmann Institute for Molecular Life Sciences, Goethe Universita¨t, Frankfurt am Main, Germany ABSTRACT Several members of the genus Legionella cause Legionnaires’ disease, a potentially debilitating form of pneumonia. Studies frequently focus on the abundant number of virulence factors present in this genus. However, what is often overlooked is the role of secondary metabolites from Legionella. Following whole genome sequencing, we assembled and annotated the Legionella parisiensis DSM 19216 genome. Together with 14 other members of the Legionella, we performed comparative genomics and analysed the secondary metabolite potential of each strain. We found that Legionella contains a huge variety of biosynthetic gene clusters (BGCs) that are potentially making a significant number of novel natural products with undefined function. Surprisingly, only a single Sfp-like phosphopantetheinyl transferase is found in all Legionella strains analyzed that might be responsible for the activation of all carrier proteins in primary (fatty acid biosynthesis) and secondary metabolism (polyketide and non-ribosomal peptide synthesis). Using conserved active site motifs, we predict Submitted 29 June 2016 some novel compounds that are probably involved in cell-cell communication, Accepted 25 October 2016 Published 24 November 2016 differing to known communication systems. -

Which Organisms Are Used for Anti-Biofouling Studies
Table S1. Semi-systematic review raw data answering: Which organisms are used for anti-biofouling studies? Antifoulant Method Organism(s) Model Bacteria Type of Biofilm Source (Y if mentioned) Detection Method composite membranes E. coli ATCC25922 Y LIVE/DEAD baclight [1] stain S. aureus ATCC255923 composite membranes E. coli ATCC25922 Y colony counting [2] S. aureus RSKK 1009 graphene oxide Saccharomycetes colony counting [3] methyl p-hydroxybenzoate L. monocytogenes [4] potassium sorbate P. putida Y. enterocolitica A. hydrophila composite membranes E. coli Y FESEM [5] (unspecified/unique sample type) S. aureus (unspecified/unique sample type) K. pneumonia ATCC13883 P. aeruginosa BAA-1744 composite membranes E. coli Y SEM [6] (unspecified/unique sample type) S. aureus (unspecified/unique sample type) graphene oxide E. coli ATCC25922 Y colony counting [7] S. aureus ATCC9144 P. aeruginosa ATCCPAO1 composite membranes E. coli Y measuring flux [8] (unspecified/unique sample type) graphene oxide E. coli Y colony counting [9] (unspecified/unique SEM sample type) LIVE/DEAD baclight S. aureus stain (unspecified/unique sample type) modified membrane P. aeruginosa P60 Y DAPI [10] Bacillus sp. G-84 LIVE/DEAD baclight stain bacteriophages E. coli (K12) Y measuring flux [11] ATCC11303-B4 quorum quenching P. aeruginosa KCTC LIVE/DEAD baclight [12] 2513 stain modified membrane E. coli colony counting [13] (unspecified/unique colony counting sample type) measuring flux S. aureus (unspecified/unique sample type) modified membrane E. coli BW26437 Y measuring flux [14] graphene oxide Klebsiella colony counting [15] (unspecified/unique sample type) P. aeruginosa (unspecified/unique sample type) graphene oxide P. aeruginosa measuring flux [16] (unspecified/unique sample type) composite membranes E. -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341 -

The Risk to Human Health from Free-Living Amoebae Interaction with Legionella in Drinking and Recycled Water Systems
THE RISK TO HUMAN HEALTH FROM FREE-LIVING AMOEBAE INTERACTION WITH LEGIONELLA IN DRINKING AND RECYCLED WATER SYSTEMS Dissertation submitted by JACQUELINE MARIE THOMAS BACHELOR OF SCIENCE (HONOURS) AND BACHELOR OF ARTS, UNSW In partial fulfillment of the requirements for the award of DOCTOR OF PHILOSOPHY in ENVIRONMENTAL ENGINEERING SCHOOL OF CIVIL AND ENVIRONMENTAL ENGINEERING FACULTY OF ENGINEERING MAY 2012 SUPERVISORS Professor Nicholas Ashbolt Office of Research and Development United States Environmental Protection Agency Cincinnati, Ohio USA and School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Professor Richard Stuetz School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Doctor Torsten Thomas School of Biotechnology and Biomolecular Sciences Faculty of Science The University of New South Wales Sydney, Australia ORIGINALITY STATEMENT '1 hereby declare that this submission is my own work and to the best of my knowledge it contains no materials previously published or written by another person, or substantial proportions of material which have been accepted for the award of any other degree or diploma at UNSW or any other educational institution, except where due acknowledgement is made in the thesis. Any contribution made to the research by others, with whom 1 have worked at UNSW or elsewhere, is explicitly acknowledged in the thesis. I also declare that the intellectual content of this thesis is the product of my own work, except to the extent that assistance from others in the project's design and conception or in style, presentation and linguistic expression is acknowledged.' Signed ~ ............................ -

Host-Adaptation in Legionellales Is 2.4 Ga, Coincident with Eukaryogenesis
bioRxiv preprint doi: https://doi.org/10.1101/852004; this version posted February 27, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Host-adaptation in Legionellales is 2.4 Ga, 2 coincident with eukaryogenesis 3 4 5 Eric Hugoson1,2, Tea Ammunét1 †, and Lionel Guy1* 6 7 1 Department of Medical Biochemistry and Microbiology, Science for Life Laboratories, 8 Uppsala University, Box 582, 75123 Uppsala, Sweden 9 2 Department of Microbial Population Biology, Max Planck Institute for Evolutionary 10 Biology, D-24306 Plön, Germany 11 † current address: Medical Bioinformatics Centre, Turku Bioscience, University of Turku, 12 Tykistökatu 6A, 20520 Turku, Finland 13 * corresponding author 14 1 bioRxiv preprint doi: https://doi.org/10.1101/852004; this version posted February 27, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 15 Abstract 16 Bacteria adapting to living in a host cell caused the most salient events in the evolution of 17 eukaryotes, namely the seminal fusion with an archaeon 1, and the emergence of both the 18 mitochondrion and the chloroplast 2. A bacterial clade that may hold the key to understanding 19 these events is the deep-branching gammaproteobacterial order Legionellales – containing 20 among others Coxiella and Legionella – of which all known members grow inside eukaryotic 21 cells 3. -

Genetic and Functional Studies of the Mip Protein of Legionella
t1.ì. Genetic and Functional Studies of the Mip Protein of Legionella Rodney Mark Ratcliff, BSc (Hons)' MASM Infectious Diseases Laboratories Institute of Medical and Veterinary Science and Department of Microbiology and Immunology UniversitY of Adelaide. Adelaide, South Australia A thesis submitted to the University of Adelaide for the degree of I)octor of Philosophy 15'h March 2000 amended 14th June 2000 Colonies of several Legionella strains on charcoal yeast extract agar (CYE) after 4 days incubation at 37"C in air. Various magnifications show typical ground-glass opalescent appearance. Some pure strains exhibit pleomorphic growth or colour. The top two photographs demonstrate typical red (LH) and blue-white (RH) fluorescence exhibited by some species when illuminated by a Woods (IJV) Lamp. * t Table of Contents .1 Chapter One: Introduction .1 Background .............'. .2 Morphology and TaxonomY J Legionellosis ............. 5 Mode of transmission "..'....'. 7 Environmental habitat 8 Interactions between Legionella and phagocytic hosts 9 Attachment 11 Engulfment and internalisation.'.. 13 Intracellular processing 13 Intracellular replication of legionellae .. " "' " "' 15 Host cell death and bacterial release 18 Virulence (the Genetic factors involved with intracellular multiplication and host cell killing .20 icm/dot system) Legiolysin .25 Msp (Znn* metaloprotease) ...'..... .25 .28 Lipopolysaccharide .29 The association of flagella with disease.. .30 Type IV fimbriae.... .31 Major outer membrane proteins....'.......'. JJ Heat shock proteins'.'. .34 Macrophage infectivity potentiator (Mip) protein Virulenceiraits of Legionella species other than L. pneumophila..........' .39 phylogeny .41 Chapter One (continued): Introduction to bacterial classification and .41 Identificati on of Legionella...'.,..'.. .46 Phylogeny .52 Methods of phylogenetic analysis' .53 Parsimony methods.'.. .55 Distance methods UPGMA cluster analYsis.'.'... -

Electronic Laboratory Reporting Use Case January 2011
ILLINOIS HEALTH INFORMATION EXCHANGE Electronic Laboratory Reporting Use Case Electronic Laboratory Reporting and Health Information Exchange Illinois Health Information Exchange Public Health Work Group January 2011 Electronic Laboratory Reporting Use Case January 2011 Table of Contents 1.0 Executive Summary……………………………………………………………………….3 2.0 Introduction…………………………………………………………………...……………..5 3.0 Scope……………………………………..………………………………………………………5 4.0 Use Case Stakeholders…………………………………………………………….….....6 5.0 Issues and Obstacles……………………………………………………………………...8 6.0 Use Case Pre-Conditions .………………….…………………………………………...8 7.0 Use Case Post-Conditions.……………………………………………………………...9 8.0 Detailed Scenarios/Technical Specifications.………………………………10 9.0 Validation and Certification………………………………………………………...12 Appendix ………………………………………………………………………………………….....13 Page 2 Electronic Laboratory Reporting Use Case January 2011 1.0 Executive Summary This Use Case is a product of the Public Health Work Group (PHWG) of the Illinois Health Information Exchange (HIE) Advisory Committee. The Illinois HIE Advisory Committee was constituted as the diverse public healthcare stakeholder body providing input and recommendations on the creation of the Illinois Health Information Exchange Authority (“the Authority”) as the Illinois vehicle for designing and implementing electronic health information exchange in Illinois. The establishment of the Authority marks the formal transition of the work of the HIE Advisory Committee and the Work Groups into alignment with the provisions of Illinois -
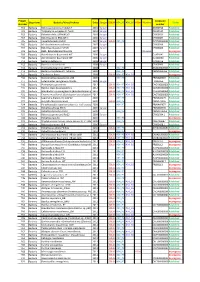
Project Number Organisms Bacteria/Virus/Archaea Date
Project_ Accession Organisms Bacteria/Virus/Archaea Date Sanger SOLiD 454_PE 454_SG PGM Illumina Status Number number P01 Bacteria Rickettsia conorii str.Malish 7 2001 Sanger AE006914 Published P02 Bacteria Tropheryma whipplei str.Twist 2003 Sanger AE014184 Published P03 Bacteria Rickettsia felis URRWXCal2 2005 Sanger CP000053 Published P04 Bacteria Rickettsia bellii RML369-C 2006 Sanger CP000087 Published P05 Bacteria Coxiella burnetii CB109 2007 Sanger SOLiD 454_PE AKYP00000000 Published P06 Bacteria Minibacterium massiliensis 2007 Sanger CP000269 Published P07 Bacteria Rickettsia massiliae MTU5 2007 Sanger CP000683 Published P08 Bacteria BaBL=Bête à Bernard Lascola 2007 Illumina In progress P09 Bacteria Acinetobacter baumannii AYE 2006 Sanger CU459141 Published P10 Bacteria Acinetobacter baumannii SDF 2006 Sanger CU468230 Published P11 Bacteria Borrelia duttonii Ly 2008 Sanger CP000976 Published P12 Bacteria Borrelia recurrentis A1 2008 Sanger CP000993 Published P13 Bacteria Francisella tularensis URFT1 2008 454_PE ABAZ00000000Published P14 Bacteria Borrelia crocidurae str. Achema 2009 454_PE PRJNA162335 Published P15 Bacteria Citrobacter koseri 2009 SOLiD 454_PE 454_SG In progress P16 Bacteria Diplorickettsia massiliensis 20B 2009 454_PE PRJNA86907 Published P17 Bacteria Enterobacter aerogenes EA1509E 2009 Sanger FO203355 Published P18 Bacteria Actinomyces grossensis 2012 SOLiD 454_PE 454_SG CAGY00000000Published P19 Bacteria Bacillus massiliosenegalensis 2012 SOLiD 454_PE 454_SG CAHJ00000000 Published P20 Bacteria Brevibacterium senegalensis -

EVALUATION of the P45 MOBILE INTEGRATIVE ELEMENT and ITS ROLE IN
EVALUATION OF THE p45 MOBILE INTEGRATIVE ELEMENT AND ITS ROLE IN Legionella pneumophila VIRULENCE A Dissertation by LANETTE M. CHRISTENSEN Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, Jeffrey D. Cirillo Committee Members, James Samuel Jon Skare Farida Sohrabji Head of Program, Warren Zimmer May 2018 Major Subject: Medical Sciences Copyright 2018 Lanette Christensen ABSTRACT Legionella pneumophila are aqueous environmental bacilli that live within protozoal species and cause a potentially fatal form of pneumonia called Legionnaires’ disease. Not all L. pneumophila strains have the same capacity to cause disease in humans. The majority of strains that cause clinically relevant Legionnaires’ disease harbor the p45 mobile integrative genomic element. Contribution of the p45 element to L. pneumophila virulence and ability to withstand environmental stress were addressed in this study. The L. pneumophila Philadelphia-1 (Phil-1) mobile integrative element, p45, was transferred into the attenuated strain Lp01 via conjugation, designating p45 an integrative conjugative element (ICE). The resulting trans-conjugate, Lp01+p45, was compared with strains Phil-1 and Lp01 to assess p45 in virulence using a guinea pig model infected via aerosol. The p45 element partially recovered the loss of virulence in Lp01 compared to that of Phil-1 evident in morbidity, mortality, and bacterial burden in the lungs at the time of death. This phenotype was accompanied by enhanced expression of type II interferon in the lungs and spleens 48 hours after infection, independent of bacterial burden. -
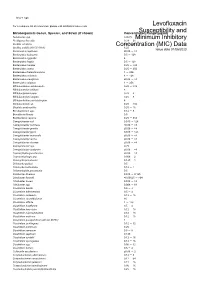
Susceptibility and Resistance Data
toku-e logo For a complete list of references, please visit antibiotics.toku-e.com Levofloxacin Microorganism Genus, Species, and Strain (if shown) Concentration Range (μg/ml)Susceptibility and Aeromonas spp. 0.0625 Minimum Inhibitory Alcaligenes faecalis 0.39 - 25 Bacillus circulans Concentration0.25 - 8 (MIC) Data Bacillus subtilis (ATCC 6051) 6.25 Issue date 01/06/2020 Bacteroides capillosus ≤0.06 - >8 Bacteroides distasonis 0.5 - 128 Bacteroides eggerthii 4 Bacteroides fragilis 0.5 - 128 Bacteroides merdae 0.25 - >32 Bacteroides ovatus 0.25 - 256 Bacteroides thetaiotaomicron 1 - 256 Bacteroides uniformis 4 - 128 Bacteroides ureolyticus ≤0.06 - >8 Bacteroides vulgatus 1 - 256 Bifidobacterium adolescentis 0.25 - >32 Bifidobacterium bifidum 8 Bifidobacterium breve 0.25 - 8 Bifidobacterium longum 0.25 - 8 Bifidobacterium pseudolongum 8 Bifidobacterium sp. 0.25 - >32 Bilophila wadsworthia 0.25 - 16 Brevibacterium spp. 0.12 - 8 Brucella melitensis 0.5 Burkholderia cepacia 0.25 - 512 Campylobacter coli 0.015 - 128 Campylobacter concisus ≤0.06 - >8 Campylobacter gracilis ≤0.06 - >8 Campylobacter jejuni 0.015 - 128 Campylobacter mucosalis ≤0.06 - >8 Campylobacter rectus ≤0.06 - >8 Campylobacter showae ≤0.06 - >8 Campylobacter spp. 0.25 Campylobacter sputorum ≤0.06 - >8 Capnocytophaga ochracea ≤0.06 - >8 Capnocytophaga spp. 0.006 - 2 Chlamydia pneumonia 0.125 - 1 Chlamydia psittaci 0.5 Chlamydia trachomatis 0.12 - 1 Chlamydophila pneumonia 0.5 Citrobacter diversus 0.015 - 0.125 Citrobacter freundii ≤0.00625 - >64 Citrobacter koseri 0.015 - -

Aquascreen® Legionella Species Qpcr Detection Kit
AquaScreen® Legionella species qPCR Detection Kit INSTRUCTIONS FOR USE FOR USE IN RESEARCH AND QUALITY CONTROL Symbols Lot No. Cat. No. Expiry date Storage temperature Number of reactions Manufacturer INDICATION The AquaScreen® Legionella species qPCR Detection kit is specifically designed for the quantitative detection of several Legionella species in water samples prepared with the AquaScreen® FastExt- ract kit. Its design complies with the requirements of AFNOR T90-471 and ISO/TS 12869:2012. Legionella are ubiquitous bacteria in surface water and moist soil, where they parasitize protozoa. The optimal growth temperature lies between +15 and +45 °C, whereas these gram-negative bacteria are dormant below 20 °C and do not survive above 60 °C. Importantly, Legionella are well-known as opportunistic intracellular human pathogens causing Legionnaires’ disease and Pontiac fever. The transmission occurs through inhalation of contami- nated aerosols generated by an infected source (e.g. human-made water systems like shower- heads, sink faucets, heaters, cooling towers, and many more). In order to efficiently prevent Legionella outbreaks, water safety control measures need syste- matic application but also reliable validation by fast Legionella testing. TEST PRINCIPLE The AquaScreen® Legionella species Kit uses qPCR for quantitative detection of legionella in wa- ter samples. In contrast to more time-consuming culture-based methods, AquaScreen® assays need less than six hours including sample preparation and qPCR to reliably detect Legionella. Moreover, the AquaScreen® qPCR assay has proven excellent performance in terms of specificity and sensitivity: other bacterial genera remain undetected whereas linear quantification is obtai- ned up to 1 x 106 particles per sample, therefore requiring no material dilution.