Severe Phenotype of Phosphorylase Kinase-Deficient Liver
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Analysis of the Human Serum Proteome
Cedarville University DigitalCommons@Cedarville Pharmaceutical Sciences Faculty Publications Department of Pharmaceutical Sciences 6-2004 Analysis of the Human Serum Proteome King C. Chan David A. Lucas Denise Hise Carl F. Schaefer Zhen Xiao See next page for additional authors Follow this and additional works at: https://digitalcommons.cedarville.edu/ pharmaceutical_sciences_publications Part of the Pharmacy and Pharmaceutical Sciences Commons This Article is brought to you for free and open access by DigitalCommons@Cedarville, a service of the Centennial Library. It has been accepted for inclusion in Pharmaceutical Sciences Faculty Publications by an authorized administrator of DigitalCommons@Cedarville. For more information, please contact [email protected]. Authors King C. Chan, David A. Lucas, Denise Hise, Carl F. Schaefer, Zhen Xiao, George M. Janini, Kenneth H. Buetow, Haleem J. Issaq, Timothy D. Veenstra, and Thomas P. Conrads Clinical Proteomics Journal Copyright ©Humana Press Inc. All rights of any nature whatsoever are reserved. ISSN 1542-6416/04/01:101–225/$25.00 Serum/Plasma Proteome Analysis of the Human Serum Proteome King C. Chan,1,† David A. Lucas,1,† Denise Hise,2 Carl F. Schaefer,2 Zhen Xiao,1 George M. Janini,1 Kenneth H. Buetow,2 Haleem J. Issaq,1 Timothy D.Veenstra,1 and Thomas P. Conrads1,* 1Laboratory of Proteomics and Analytical Technologies, National Cancer Institute at Frederick, SAIC-Frederick, Inc, PO Box B, Frederick, MD 21702 2Center for Bioinformatics, National Cancer Institute, Bethesda, MD 20892 †These authors contributed equally to this work. each of which was analyzed by microcapillary Abstract reversed-phase liquid chromatography coupled Changes in serum proteins that signal online with MS/MS analysis. -
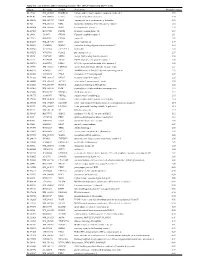
Supplementary Table 1
Table S1. List of Genes Differentially Expressed in YB-1 siRNA-Transfected MCF-7 Cells Unigene Accession Symbol Description Mean fold change Hs.17466 NM_004585 RARRES3 retinoic acid receptor responder (tazarotene induced) 3 3.57 Hs.64746 NM_004669 CLIC3 chloride intracellular channel 3 3.33 Hs.516155 NM_001747 CAPG capping protein (actin filament), gelsolin-like 2.88 Hs.926 NM_002463 MX2 myxovirus (influenza virus) resistance 2 (mouse) 2.72 Hs.643494 NM_005410 SEPP1 selenoprotein P, plasma, 1 2.70 Hs.267038 BC017500 POF1B premature ovarian failure 1B 2.67 Hs.20961 U63917 GPR30 G protein- coupled receptor 30 2532.53 Hs.199877 BE645967 CPNE4 copine IV 2.49 Hs.632177 NM_017458 MVP major vault protein 2.48 Hs.528836 AA005023 NOD27 nucleotide-binding oligomerization domains 27 2.43 Hs.360940 AL589866 dJ222E13.1 kraken-like 2.40 Hs.515575 AI743780 PLAC2 placenta-specific 2 2.37 Hs.25674 AI827820 MBD2 methyl-CpG binding domain protein 2 2.36 Hs.12229 AA149594 TIEG2 TGFB inducible early growth response 2 2.33 Hs.482730 AA053711 EDIL3 EGF-like repeats and discoidin I-like domains 3 2.32 Hs. 370771 NM_000389 CDKN1A cyclin- depen dent kinase in hibitor 1A (p 21, Cip 1) 2282.28 Hs.469175 AI341537 JFC1 NADPH oxidase-related, C2 domain-containing protein 2.28 Hs.514821 AF043341 CCL5 chemokine (C-C motif) ligand 5 2.27 Hs.591292 NM_023915 GPR87 G protein-coupled receptor 87 2.26 Hs.500483 NM_001613 ACTA2 actin, alpha 2, smooth muscle, aorta 2.24 Hs.632824 NM_006729 DIAPH2 diaphanous homolog 2 (Drosophila) 2.22 Hs.369430 NM_000919 PAM peptidylglycine -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

PHKG2, Active (SRP5062)
PHKG2, active, GST tagged, human PRECISIOÒ Kinase recombinant, expressed in Sf9 cells Catalog Number SRP5062 Storage Temperature –70°C Synonyms: GSD9C Figure 1. SDS-PAGE Gel of Typical Lot Product Description 70–95% (densitometry) PHKG2 is the hepatic and testis isoform of the gamma subunit of phosphorylase kinase. PHKG2 gene contains 10 exons and spans 9.5 kb and maps to chromosome 16p12.1-p11.2.1 Deficiency of PHK, a regulatory enzyme of glycogen metabolism, is responsible for 25% of all cases of glycogen storage disease and is genetically and clinically heterogeneous. Mutations in the PHKG2 gene lead to autosomal liver-specific PHK deficiency (glycogen storage disease IXc) and an increased risk of cirrhosis, and at least 11 PHKG2 mutations have been identified to date.2 Figure 2. Specific Activity of Typical Lot Recombinant, full-length, human PHKG2 was 59–81 nmole/min/mg expressed by baculovirus in Sf9 insect cells using an N-terminal GST tag. The gene accession number is NM_000294. Recombinant protein stored in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.25 mM DTT, 0.1 mM EDTA, 0.1 mM PMSF, and 25% glycerol. Molecular mass: ~70 kDa Purity: 70–95% (SDS-PAGE, see Figure 1) Specific Activity: 59–81 nmole/min/mg (see Figure 2) Precautions and Disclaimer Procedure This product is for R&D use only, not for drug, Preparation Instructions household, or other uses. Please consult the Material Kinase Assay Buffer – 25 mM MOPS, pH 7.2, 12.5 mM Safety Data Sheet for information regarding hazards glycerol 2-phosphate, 25 mM MgCl2, 5 mM EGTA, and and safe handling practices. -

Supplementary Table 1. in Vitro Side Effect Profiling Study for LDN/OSU-0212320. Neurotransmitter Related Steroids
Supplementary Table 1. In vitro side effect profiling study for LDN/OSU-0212320. Percent Inhibition Receptor 10 µM Neurotransmitter Related Adenosine, Non-selective 7.29% Adrenergic, Alpha 1, Non-selective 24.98% Adrenergic, Alpha 2, Non-selective 27.18% Adrenergic, Beta, Non-selective -20.94% Dopamine Transporter 8.69% Dopamine, D1 (h) 8.48% Dopamine, D2s (h) 4.06% GABA A, Agonist Site -16.15% GABA A, BDZ, alpha 1 site 12.73% GABA-B 13.60% Glutamate, AMPA Site (Ionotropic) 12.06% Glutamate, Kainate Site (Ionotropic) -1.03% Glutamate, NMDA Agonist Site (Ionotropic) 0.12% Glutamate, NMDA, Glycine (Stry-insens Site) 9.84% (Ionotropic) Glycine, Strychnine-sensitive 0.99% Histamine, H1 -5.54% Histamine, H2 16.54% Histamine, H3 4.80% Melatonin, Non-selective -5.54% Muscarinic, M1 (hr) -1.88% Muscarinic, M2 (h) 0.82% Muscarinic, Non-selective, Central 29.04% Muscarinic, Non-selective, Peripheral 0.29% Nicotinic, Neuronal (-BnTx insensitive) 7.85% Norepinephrine Transporter 2.87% Opioid, Non-selective -0.09% Opioid, Orphanin, ORL1 (h) 11.55% Serotonin Transporter -3.02% Serotonin, Non-selective 26.33% Sigma, Non-Selective 10.19% Steroids Estrogen 11.16% 1 Percent Inhibition Receptor 10 µM Testosterone (cytosolic) (h) 12.50% Ion Channels Calcium Channel, Type L (Dihydropyridine Site) 43.18% Calcium Channel, Type N 4.15% Potassium Channel, ATP-Sensitive -4.05% Potassium Channel, Ca2+ Act., VI 17.80% Potassium Channel, I(Kr) (hERG) (h) -6.44% Sodium, Site 2 -0.39% Second Messengers Nitric Oxide, NOS (Neuronal-Binding) -17.09% Prostaglandins Leukotriene, -

Phosphoproteomics of Retinoblastoma: a Pilot Study Identifies Aberrant Kinases
molecules Article Phosphoproteomics of Retinoblastoma: A Pilot Study Identifies Aberrant Kinases Lakshmi Dhevi Nagarajha Selvan 1,†, Ravikanth Danda 1,2,†, Anil K. Madugundu 3 ID , Vinuth N. Puttamallesh 3, Gajanan J. Sathe 3,4, Uma Maheswari Krishnan 2, Vikas Khetan 5, Pukhraj Rishi 5, Thottethodi Subrahmanya Keshava Prasad 3,6 ID , Akhilesh Pandey 3,7,8, Subramanian Krishnakumar 1, Harsha Gowda 3,* and Sailaja V. Elchuri 9,* 1 L&T Opthalmic Pathology, Vision Research Foundation, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India; [email protected] (L.D.N.S.); [email protected] (R.D.); [email protected] (S.K.) 2 Centre for Nanotechnology and Advanced Biomaterials, Shanmugha Arts, Science, Technology and Research Academy University, Tanjore, Tamil Nadu 613 401, India; [email protected] 3 Institute of Bioinformatics, International Technology Park, Bangalore, Karnataka 560 066, India; [email protected] (A.K.M.); [email protected] (V.N.P.); [email protected] (G.J.S.); [email protected] (T.S.K.P.); [email protected] (A.P.) 4 Manipal Academy of Higher Education (MAHE), Manipal, Karnataka 576 104, India 5 Shri Bhagwan Mahavir Vitreoretinal Services, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India; [email protected] (V.K.); [email protected] (P.R.) 6 Center for Systems Biology and Molecular Medicine, Yenepoya Research Centre, Yenepoya (Deemed to be University), Mangalore, Karnataka 575 108, India 7 McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 8 Departments of Biological Chemistry, Pathology and Oncology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 9 Department of Nanotechnology, Vision Research Foundation, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India * Correspondence: [email protected] (H.G.); [email protected] (S.V.E.); Tel.: +91-80-28416140 (H.G.); +91-44-28271616 (S.V.E.) † These authors contributed equally to this work. -

Prognostic and Immune Implications of a Novel Ferroptosis-Related Ten-Gene Signature in Lung Adenocarcinoma
1058 Original Article Page 1 of 26 Prognostic and immune implications of a novel ferroptosis-related ten-gene signature in lung adenocarcinoma Chao Ma1,2^, Feng Li1,3, Huan Luo1 1Charité – Universitätsmedizin Berlin, Freie Universität Berlin, Humboldt-Universität zu Berlin, Berlin Institute of Health, Berlin, Germany; 2Berlin Institute of Health Center for Regenerative Therapies and Berlin-Brandenburg Center for Regenerative Therapies (BCRT), Charité – Universitätsmedizin Berlin, Berlin, Germany; 3Department of Surgery, Competence Center of Thoracic Surgery, Charité University Hospital Berlin, Berlin, Germany Contributions: (I) Conception and design: C Ma; (II) Administrative support: C Ma; (III) Provision of study materials or patients: C Ma, F Li; (IV) Collection and assembly of data: All authors; (V) Data analysis and interpretation: C Ma; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Chao Ma. Berlin Institute of Health Center for Regenerative Therapies and Berlin-Brandenburg Center for Regenerative Therapies (BCRT), Charité – Universitätsmedizin Berlin, Berlin, Germany. Email: [email protected]. Background: Lung cancer has been the focus of attention for many researchers in recent years due to its leading contribution to cancer-related death worldwide, with lung adenocarcinoma (LUAD) being the most common histological type. Ferroptosis, a novel iron-dependent form of regulated cell death, can be induced by sorafenib. Emerging evidence shows that triggering ferroptosis has potential as a cancer therapy. This work aimed to build a ferroptosis-related gene signature for predicting the outcome of LUAD. Methods: The TCGA-LUAD dataset was set as the training cohort, and the GSE72094 and GSE68465 datasets were set as the validation cohorts. -

Full Disclosure Forms
Expanding genotype/phenotype of neuromuscular diseases by comprehensive target capture/NGS Xia Tian, PhD* ABSTRACT * Wen-Chen Liang, MD Objective: To establish and evaluate the effectiveness of a comprehensive next-generation * Yanming Feng, PhD sequencing (NGS) approach to simultaneously analyze all genes known to be responsible for Jing Wang, MD the most clinically and genetically heterogeneous neuromuscular diseases (NMDs) involving spi- Victor Wei Zhang, PhD nal motoneurons, neuromuscular junctions, nerves, and muscles. Chih-Hung Chou, MS Methods: All coding exons and at least 20 bp of flanking intronic sequences of 236 genes causing Hsien-Da Huang, PhD NMDs were enriched by using SeqCap EZ solution-based capture and enrichment method fol- Ching Wan Lam, PhD lowed by massively parallel sequencing on Illumina HiSeq2000. Ya-Yun Hsu, PhD ; 3 Thy-Sheng Lin, MD Results: The target gene capture/deep sequencing provides an average coverage of 1,000 per Wan-Tzu Chen, MS nucleotide. Thirty-five unrelated NMD families (38 patients) with clinical and/or muscle pathologic Lee-Jun Wong, PhD diagnoses but without identified causative genetic defects were analyzed. Deleterious mutations Yuh-Jyh Jong, MD were found in 29 families (83%). Definitive causative mutations were identified in 21 families (60%) and likely diagnoses were established in 8 families (23%). Six families were left without diagnosis due to uncertainty in phenotype/genotype correlation and/or unidentified causative Correspondence to genes. Using this comprehensive panel, we not only identified mutations in expected genes but Dr. Wong: also expanded phenotype/genotype among different subcategories of NMDs. [email protected] or Dr. Jong: Conclusions: Target gene capture/deep sequencing approach can greatly improve the genetic [email protected] diagnosis of NMDs. -

Muscle Glycogen Phosphorylase and Its Functional Partners in Health and Disease
cells Review Muscle Glycogen Phosphorylase and Its Functional Partners in Health and Disease Marta Migocka-Patrzałek * and Magdalena Elias Department of Animal Developmental Biology, Faculty of Biological Sciences, University of Wroclaw, 50-335 Wroclaw, Poland; [email protected] * Correspondence: [email protected] Abstract: Glycogen phosphorylase (PG) is a key enzyme taking part in the first step of glycogenolysis. Muscle glycogen phosphorylase (PYGM) differs from other PG isoforms in expression pattern and biochemical properties. The main role of PYGM is providing sufficient energy for muscle contraction. However, it is expressed in tissues other than muscle, such as the brain, lymphoid tissues, and blood. PYGM is important not only in glycogen metabolism, but also in such diverse processes as the insulin and glucagon signaling pathway, insulin resistance, necroptosis, immune response, and phototransduction. PYGM is implicated in several pathological states, such as muscle glycogen phosphorylase deficiency (McArdle disease), schizophrenia, and cancer. Here we attempt to analyze the available data regarding the protein partners of PYGM to shed light on its possible interactions and functions. We also underline the potential for zebrafish to become a convenient and applicable model to study PYGM functions, especially because of its unique features that can complement data obtained from other approaches. Keywords: PYGM; muscle glycogen phosphorylase; functional protein partners; glycogenolysis; McArdle disease; cancer; schizophrenia Citation: Migocka-Patrzałek, M.; Elias, M. Muscle Glycogen Phosphorylase and Its Functional Partners in Health and Disease. Cells 1. Introduction 2021, 10, 883. https://doi.org/ The main energy substrate in animal tissues is glucose, which is stored in the liver and 10.3390/cells10040883 muscles in the form of glycogen, a polymer consisting of glucose molecules.