Structure-Guided Microbial Targeting of Antistaphylococcal Prodrugs
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Antibiotic Practice Change to Curtail Linezolid Use in Pediatric Hospitalized Patients in Hawai‘I with Uncomplicated Skin and Soft Tissue Infections
Antibiotic Practice Change to Curtail Linezolid Use in Pediatric Hospitalized Patients in Hawai‘i with Uncomplicated Skin and Soft Tissue Infections Cheryl Okado MD and Tori Teramae BS Abstract MRSA coverage, clindamycin became a widely-used antibi- otic for treating uncomplicated SSTIs in children. Following Antimicrobial resistance affects health care providers’ choice of antibiotics increasing clindamycin use, increasing clindamycin resistance in the treatment of skin and soft tissue infections (SSTIs). Based on local was soon noted, particularly in MRSA isolates.2 Prior to 2010, antibiotic susceptibility data showing high clindamycin resistance and high MRSA predominance appeared to peak, and since then it has MRSA prevalence, a change in antibiotic regimen for children hospitalized for 3 uncomplicated SSTIs was instituted in an attempt to curb the use of linezolid. been decreasing. The prevalence of clindamycin-resistant GAS A retrospective chart review was performed on 278 pediatric patients with has been known to vary with time and location with US rates uncomplicated SSTIs hospitalized at Kapi‘olani Medical Center for Women ranging from 4%-41% since the early 2000s.4 and Children in Hawai‘i from May 2014 to April 2015 and November 2015 to October 2016. Data consisted of 12 months of baseline data and 12 In Hawai‘i, methicillin and clindamycin resistance patterns of months of data post-implementation of an antibiotic combination regimen of SA initially followed similar increasing trends. Antibiograms 2 widely-used antibiotics: high-dose cefazolin and high-dose clindamycin. at Hawai‘i’s children’s hospital, Kapi‘olani Medical Center for Practitioners were encouraged to use cefazolin alone if clinical suspicion was high for single-organism infection with group A streptococcus. -

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Isolation and Characterization of the Prolyl Aminopeptidase Gene (Pap) from Aeromonas Sobria: Comparison with the Bacillus Coagulans Enzyme1
J. Biochem. 116, 818-825 (1994) Isolation and Characterization of the Prolyl Aminopeptidase Gene (pap) from Aeromonas sobria: Comparison with the Bacillus coagulans Enzyme1 Ana Kitazono,* Atsuko Kitano,* Daisuke Tsuru,•õ and Tadashi Yoshimoto*,2 *School of Pharmaceutical Sciences , Nagasaki University, 1-14 Bunkyo-machi, Nagasaki, Nagasaki 852; and •õ Department of Applied Microbiology, Kumamoto Institute of Technology, 4-22-1 Ikeda, Kumamoto, Kumamoto 860 Received for publication, May 16, 1994 The Aeromonas sobria pap gene encoding prolyl aminopeptidase (PAP) was cloned. It consists of 425 codons and encodes a homotetrameric enzyme of 205kDa. The purified enzyme showed an almost absolute specificity for amino-terminal proline. Proline and hydroxyproline residues from many peptide and amide substrates could be easily removed, while no activity was detected for substrates having other amino terminals. The enzyme was very similar to that from Bacillus coagulans in many aspects, such as the strong inhibition caused by PCMB and the weak or no inhibition caused by DFP and chelators, respectively. However, these enzymes show only 15% identity in their amino acid sequences. Differences were also observed in their molecular weight, stability and activity toward some peptide substrates. When aligning the deduced amino acid sequence with known sequences from other microorganisms, conserved sequences were found at the amino-terminal region; the significance of these conserved regions is discussed. Based on the results of this work, and on the studies available to date, the occurrence of at least two types of PAPs is postulated. One group would be formed by the Bacillus, Neisseria, and Lactobacillus enzymes, and the other by enzymes such as the Aeromonas PAP. -

P-Glycoprotein, CYP3A, and Plasma Carboxylesterase Determine Brain and Blood Disposition of the Mtor Inhibitor Everolimus (Afinitor) in Mice
Published OnlineFirst April 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1759 Clinical Cancer Cancer Therapy: Preclinical Research P-Glycoprotein, CYP3A, and Plasma Carboxylesterase Determine Brain and Blood Disposition of the mTOR Inhibitor Everolimus (Afinitor) in Mice Seng Chuan Tang1, Rolf W. Sparidans3, Ka Lei Cheung4, Tatsuki Fukami5, Selvi Durmus1, Els Wagenaar1, Tsuyoshi Yokoi5, Bart J.M. van Vlijmen4, Jos H. Beijnen2,3, and Alfred H. Schinkel1 Abstract Purpose: To clarify the role of ABCB1, ABCG2, and CYP3A in blood and brain exposure of everolimus using knockout mouse models. À À À À À À À À Experimental Design: We used wild-type, Abcb1a/1b / , Abcg2 / , Abcb1a/1b;Abcg2 / , and Cyp3a / mice to study everolimus oral bioavailability and brain accumulation. Results: Following everolimus administration, brain concentrations and brain-to-liver ratios were À À À À À À substantially increased in Abcb1a/1b / and Abcb1a/1b;Abcg2 / , but not Abcg2 / mice. The fraction of everolimus located in the plasma compartment was highly increased in all knockout strains. In vitro, everolimus was rapidly degraded in wild-type but not knockout plasma. Carboxylesterase 1c (Ces1c), a plasma carboxylesterase gene, was highly upregulated (80-fold) in the liver of knockout mice relative to wild-type mice, and plasma Ces1c likely protected everolimus from degradation by binding and stabilizing it. This binding was prevented by preincubation with the carboxylesterase inhibitor BNPP. In vivo knockdown experiments confirmed the involvement of Ces1c in everolimus stabilization. Everolimus also markedly inhibited the hydrolysis of irinotecan and p-nitrophenyl acetate by mouse plasma carboxylesterase À À and recombinant human CES2, respectively. -

Drug News Nov Issue 25
D r u g N e w s 藥 物 情 報 Issue No. 25 : November 2011 This is a monthly digest of local and overseas drug safety news and information released in the previous month. For the latest news and information, please refer to public announcements or the website of the Drug Office of the Department of Health (http://www.drugoffice.gov.hk). Safety Update US: Updated information about drug Canada and FDA released similar alerts on the interaction of Zyvox (linezolid) and interaction between methylene blue and serotonin Methylene Blue with serotonergic reuptake inhibitors. At the same time, FDA also released alert on the interaction between linezolid psychiatric medications and serotonin reuptake inhibitors. The news had 20 October 2011 – The Food and Drug been reported in Issue No. 17 and 22 of Drug News Administration (FDA) provided additional respectively. In addition, the Department of Health information about the possible serotonin syndrome (DH) issued press statement on 18 February 2011 in patients taking serotonergic psychiatric and letters were also sent to healthcare professionals medications and linezolid/ methylene blue. It was in February 2011 and July 2011. found that not all serotonergic psychiatric drugs had The product certificate holders of the an equal capacity to cause serotonin syndrome with pharmaceutical products containing methylene blue, use of linezolid/ methylene blue. According to the linezolid and serotonergic psychiatric medications FDA's Adverse Event Reporting System (AERS), have been requested to update the sales pack and/or most cases of serotonin syndrome with linezolid or package insert of these products to include methylene blue occurred in patients taking specific information about the potential drug interactions. -

Treatment of Drug-Resistant Tuberculosis an Official ATS/CDC/ERS/IDSA Clinical Practice Guideline Payam Nahid, Sundari R
AMERICAN THORACIC SOCIETY DOCUMENTS Treatment of Drug-Resistant Tuberculosis An Official ATS/CDC/ERS/IDSA Clinical Practice Guideline Payam Nahid, Sundari R. Mase, Giovanni Battista Migliori, Giovanni Sotgiu, Graham H. Bothamley, Jan L. Brozek, Adithya Cattamanchi, J. Peter Cegielski, Lisa Chen, Charles L. Daley, Tracy L. Dalton, Raquel Duarte, Federica Fregonese, C. Robert Horsburgh, Jr., Faiz Ahmad Khan, Fayez Kheir, Zhiyi Lan, Alfred Lardizabal, Michael Lauzardo, Joan M. Mangan, Suzanne M. Marks, Lindsay McKenna, Dick Menzies, Carole D. Mitnick, Diana M. Nilsen, Farah Parvez, Charles A. Peloquin, Ann Raftery, H. Simon Schaaf, Neha S. Shah, Jeffrey R. Starke, John W. Wilson, Jonathan M. Wortham, Terence Chorba, and Barbara Seaworth; on behalf of the American Thoracic Society, U.S. Centers for Disease Control and Prevention, European Respiratory Society, and Infectious Diseases Society of America THIS OFFICIAL CLINICAL PRACTICE GUIDELINE WAS APPROVED BY THE AMERICAN THORACIC SOCIETY, THE EUROPEAN RESPIRATORY SOCIETY, AND THE INFECTIOUS DISEASES SOCIETY OF AMERICA SEPTEMBER 2019, AND WAS CLEARED BY THE U.S. CENTERS FOR DISEASE CONTROL AND PREVENTION SEPTEMBER 2019 Background: The American Thoracic Society, U.S. Centers for was judged to be very low, because the data came Disease Control and Prevention, European Respiratory Society, and from observational studies with significant loss to follow-up Infectious Diseases Society of America jointly sponsored this new and imbalance in background regimens between comparator practice guideline on the treatment of drug-resistant tuberculosis groups. Good practices in the management of MDR-TB are (DR-TB). The document includes recommendations on the described. On the basis of the evidence review, a clinical strategy treatment of multidrug-resistant TB (MDR-TB) as well as tool for building a treatment regimen for MDR-TB is also isoniazid-resistant but rifampin-susceptible TB. -
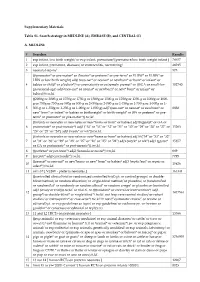
EMBASE (B), and CENTRAL (C) A. MEDLINE # Searches
Supplementary Materials Table S1. Search strategy in MEDLINE (A), EMBASE (B), and CENTRAL (C) A. MEDLINE # Searches Results 1 exp infant, low birth weight/ or exp infant, premature/ [premature/low birth weight infant ] 78657 2 exp infant, premature, diseases/ or enterocolitis, necrotizing/ 46015 3 neonatal sepsis/ 575 (((prematur* or pre-matur* or i?matur* or preterm* or pre-term* or VLBW* or ELBW* or LBW or low birth weight) adj6 (neo-nat* or neonat* or newborn* or born* or infant* or 4 babies or child* or p?ediatr*)) or prematurity or extremely premat* or ((SGA or small-for- 102740 gestational-age) adj6 (neo-nat* or neonat* or newborn* or new* born* or infant* or babies))).tw,kf. ((2000g or 2000-g or 1750g or 1750-g or 1500g or 1500-g or 1250g or 1250-g or 1000g or 1000- g or 750g or 750-g or 500g or 500-g or 2-000g or 2-000-g or 1-750g or 1-750-g or 1-500g or 1- 5 500-g or 1-250g or 1-250-g or 1-000g or 1-000-g) adj7 (neo-nat* or neonat* or newborn* or 8838 new* born* or infant* or babies or birthweight* or birth weight* or BW or preterm* or pre- term* or prematur* or pre-matur*)).tw,kf. ((infants or neonates or neo-nates or new*borns or born* or babies) adj18 (gestat* or GA or 6 postmenstr* or post-menstr*) adj3 ("34" or "33" or "32" or "31" or "30" or "29" or "28" or "27" or 15263 "26" or "25" or "24") adj3 (week* or wk*)).tw,kf. -

Frequently Asked Questions on The
Frequently asked questions on the WHO treatment guidelines for isoniazid-resistant tuberculosis Version: 24 April 2018 The advice in this document has been prepared by the Global TB Programme of the World Health Organization (WHO) and is to be read alongside the WHO treatment guidelines for isoniazid-resistant tuberculosis and its online annexes. See Further reading at end for additional resources. Who are the new WHO treatment guidelines for isoniazid-resistant tuberculosis intended for ? The WHO treatment guidelines for isoniazid-resistant tuberculosis are targeted at health care professionals (doctors, nurses) and other staff involved in TB care in both low and high resource settings. The possibility of drug-resistant disease now needs to be entertained whenever treating TB patients. These guidelines provide technical detail which is helpful in making the best-informed decision in clinical practice and programme management. The answers to the frequently asked questions (FAQ) in this document intend to help implementers with common, practical issues that arise when adopting the new guidance. This document complements the WHO guidelines by elaborating further on certain aspects of implementation that were beyond the scope of the guideline itself. What are the main recommendations of these new guidelines ? The new guidelines recommend that patients with confirmed isoniazid-resistant and rifampicin susceptible tuberculosis (abbreviated to Hr-TB) are treated for 6 months with a regimen composed of rifampicin (R), ethambutol (E), pyrazinamide (Z) and levofloxacin (Lfx). The exclusion of rifampicin resistance ahead of start of this regimen is critical. It is further recommended not to add streptomycin (S) or other injectable agents to the treatment regimen. -

Medications to Be Avoided Or Used with Caution in Parkinson's Disease
Medications To Be Avoided Or Used With Caution in Parkinson’s Disease This medication list is not intended to be complete and additional brand names may be found for each medication. Every patient is different and you may need to take one of these medications despite caution against it. Please discuss your particular situation with your physician and do not stop any medication that you are currently taking without first seeking advice from your physician. Most medications should be tapered off and not stopped suddenly. Although you may not be taking these medications at home, one of these medications may be introduced while hospitalized. If a hospitalization is planned, please have your neurologist contact your treating physician in the hospital to advise which medications should be avoided. Medications to be avoided or used with caution in combination with Selegiline HCL (Eldepryl®, Deprenyl®, Zelapar®), Rasagiline (Azilect®) and Safinamide (Xadago®) Medication Type Medication Name Brand Name Narcotics/Analgesics Meperidine Demerol® Tramadol Ultram® Methadone Dolophine® Propoxyphene Darvon® Antidepressants St. John’s Wort Several Brands Muscle Relaxants Cyclobenzaprine Flexeril® Cough Suppressants Dextromethorphan Robitussin® products, other brands — found as an ingredient in various cough and cold medications Decongestants/Stimulants Pseudoephedrine Sudafed® products, other Phenylephrine brands — found as an ingredient Ephedrine in various cold and allergy medications Other medications Linezolid (antibiotic) Zyvox® that inhibit Monoamine oxidase Phenelzine Nardil® Tranylcypromine Parnate® Isocarboxazid Marplan® Note: Additional medications are cautioned against in people taking Monoamine oxidase inhibitors (MAOI), including other opioids (beyond what is mentioned in the chart above), most classes of antidepressants and other stimulants (beyond what is mentioned in the chart above). -

The Role of the B-Type Phospholipases in S. Cerevisiae: Function, Regulation, and Physiological Relevance in Lipid Homeostasis Beth a Surlow
Duquesne University Duquesne Scholarship Collection Electronic Theses and Dissertations Summer 2014 The Role of the B-Type Phospholipases in S. cerevisiae: Function, Regulation, and Physiological Relevance in Lipid Homeostasis Beth A Surlow Follow this and additional works at: https://dsc.duq.edu/etd Recommended Citation Surlow, B. (2014). The Role of the B-Type Phospholipases in S. cerevisiae: Function, Regulation, and Physiological Relevance in Lipid Homeostasis (Doctoral dissertation, Duquesne University). Retrieved from https://dsc.duq.edu/etd/1255 This Immediate Access is brought to you for free and open access by Duquesne Scholarship Collection. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Duquesne Scholarship Collection. For more information, please contact [email protected]. THE ROLE OF B-TYPE PHOSPHOLIPASES IN S. CEREVISIAE: FUNCTION, REGULATION, AND PHYSIOLOGICAL RELEVANCE IN LIPID HOMEOSTASIS A Dissertation Submitted to the Bayer School of Natural and Environmental Sciences Duquesne University In partial fulfillment of the requirements for the degree of Doctor of Philosophy By Beth A. Surlow August 2014 Copyright by Beth A. Surlow 2014 THE ROLE OF B-TYPE PHOSPHOLIPASES IN S. CEREVISIAE: FUNCTION, REGULATION, AND PHYSIOLOGICAL RELEVANCE IN LIPID HOMEOSTASIS By Beth A. Surlow Approved June 5, 2014 ________________________________ ________________________________ Dr. Jana Patton-Vogt Dr. Philip Auron Associate Professor of Biological Professor of Biological Sciences Sciences (Committee Member) (Committee Chair) ________________________________ ________________________________ Dr. Joseph McCormick Dr. Jeffrey Brodsky Chair and Associate Professor of Professor of Biological Sciences Biological Sciences University of Pittsburgh (Committee Member) (Committee Member) ________________________________ Dr. Philip Reeder Dean, Bayer School of Natural and Environmental Science iii ABSTRACT THE ROLE OF B-TYPE PHOSPHOLIPASES IN S. -

Metabolic Regulation by Lipid Activated Receptors by Maxwell A
Metabolic Regulation by Lipid Activated Receptors By Maxwell A Ruby A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy In Molecular & Biochemical Nutrition In the Graduate Division Of the University of California, Berkeley Committee in charge: Professor Marc K. Hellerstein, Chair Professor Ronald M. Krauss Professor George A. Brooks Professor Andreas Stahl Fall 2010 Abstract Metabolic Regulation by Lipid Activated Receptors By Maxwell Alexander Ruby Doctor of Philosophy in Molecular & Biochemical Nutrition University of California, Berkeley Professor Marc K. Hellerstein, Chair Obesity and related metabolic disorders have reached epidemic levels with dire public health consequences. Efforts to stem the tide focus on behavioral and pharmacological interventions. Several hypolipidemic pharmaceutical agents target endogenous lipid receptors, including the peroxisomal proliferator activated receptor α (PPAR α) and cannabinoid receptor 1 (CB1). To further the understanding of these clinically relevant receptors, we elucidated the biochemical basis of PPAR α activation by lipoprotein lipolysis products and the metabolic and transcriptional responses to elevated endocannabinoid signaling. PPAR α is activated by fatty acids and their derivatives in vitro. While several specific pathways have been implicated in the generation of PPAR α ligands, we focused on lipoprotein lipase mediated lipolysis of triglyceride rich lipoproteins. Fatty acids activated PPAR α similarly to VLDL lipolytic products. Unbound fatty acid concentration determined the extent of PPAR α activation. Lipolysis of VLDL, but not physiological unbound fatty acid concentrations, created the fatty acid uptake necessary to stimulate PPAR α. Consistent with a role for vascular lipases in the activation of PPAR α, administration of a lipase inhibitor (p-407) prevented PPAR α dependent induction of target genes in fasted mice. -

Genetic and Molecular Analysis of Two New Loci Controlling Flowering in Garden Pea
Genetic and molecular analysis of two new loci controlling flowering in garden pea By A S M Mainul Hasan School of Natural Sciences Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy University of Tasmania, July 2018 Declaration of originality This thesis contains no material which has been accepted for a degree or diploma by the University or any other institution, except by way of background information and duly acknowledge in the thesis, and to the best of my knowledge and belief no material previously published or written by another person except where due acknowledgement is made in the text of the thesis, nor does the thesis contain any material that infringes copyright. Authority of access This thesis may be made available for loan. Copying and communication of any part of this thesis is prohibited for two years from the date this statement was signed; after that time limited copying and communication is permitted in accordance with the Copyright Act 1968. Date: 6-07-2018 A S M Mainul Hasan i Abstract Flowering is one of the key developmental process associated with the life cycle of plant and it is regulated by different environmental factors and endogenous cues. In the model species Arabidopsis thaliana a mobile protein, FLOWERING LOCUS T (FT) plays central role to mediate flowering time and expression of FT is regulated by photoperiod. While flowering mechanisms are well-understood in A. thaliana, knowledge about this process is limited in legume (family Fabaceae) which are the second major group of crops after cereals in satisfying the global demand for food and fodder.