Accelerated Structure-Based Design of Chemically Diverse Allosteric
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
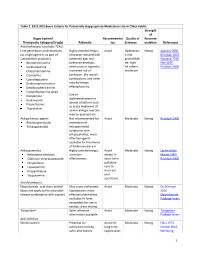
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Hypersalivation in Children and Adults
Pharmacological Management of Hypersalivation in Children and Adults Scope: Adult patients with Parkinson’s disease, children with neurodisability, cerebral palsy, long-term ventilation with drooling, and drug-induced hypersalivation. ASSESSMENT OF SEVERITY/RESPONSE TO TREATMENT: Severity of drooling can be assessed subjectively via discussion with patients and their carers/parents and by observation. Amount of drooling can be quantified by measuring the number of bibs required per day and this can also be graded using the Thomas-Stonell and Greenberg scale: • 1 = Dry (no drooling) • 2 = Mild (moist lips) • 3 = Moderate (wet lips and chin) • 4 = Severe (damp clothing) CONSIDERATIONS FOR PRESCRIBING/TITRATION No evidence to support the use of one particular treatment over another. Drug choice is to be determined by individual patient factors. When prescribing/titrating antimuscarinic drugs to treat hypersalivation always take account of: • Coexisting conditions (for example, history of urinary retention, constipation, glaucoma, dental issues, reflux etc.) • Use of other existing medication affecting the total antimuscarinic burden • Risk of adverse effects Titrate dose upward until the desired level of dryness, side effects or maximum dose reached. Take into account the preferences of the patients and their carers/ parents, and the age range and indication covered by the marketing authorisations (see individual summaries of product characteristics, BNF or BNFc for full prescribing information). FIRST LINE DRUG TREATMENT OPTIONS FOR ADULTS -
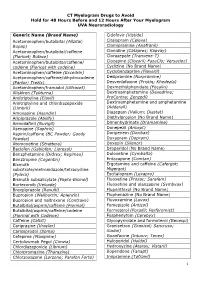
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology Generic Name (Brand Name) Cidofovir (Vistide) Acetaminophen/butalbital (Allzital; Citalopram (Celexa) Bupap) Clomipramine (Anafranil) Acetaminophen/butalbital/caffeine Clonidine (Catapres; Kapvay) (Fioricet; Butace) Clorazepate (Tranxene-T) Acetaminophen/butalbital/caffeine/ Clozapine (Clozaril; FazaClo; Versacloz) codeine (Fioricet with codeine) Cyclizine (No Brand Name) Acetaminophen/caffeine (Excedrin) Cyclobenzaprine (Flexeril) Acetaminophen/caffeine/dihydrocodeine Desipramine (Norpramine) (Panlor; Trezix) Desvenlafaxine (Pristiq; Khedezla) Acetaminophen/tramadol (Ultracet) Dexmethylphenidate (Focalin) Aliskiren (Tekturna) Dextroamphetamine (Dexedrine; Amitriptyline (Elavil) ProCentra; Zenzedi) Amitriptyline and chlordiazepoxide Dextroamphetamine and amphetamine (Limbril) (Adderall) Amoxapine (Asendin) Diazepam (Valium; Diastat) Aripiprazole (Abilify) Diethylpropion (No Brand Name) Armodafinil (Nuvigil) Dimenhydrinate (Dramamine) Asenapine (Saphris) Donepezil (Aricept) Aspirin/caffeine (BC Powder; Goody Doripenem (Doribax) Powder) Doxapram (Dopram) Atomoxetine (Strattera) Doxepin (Silenor) Baclofen (Gablofen; Lioresal) Droperidol (No Brand Name) Benzphetamine (Didrex; Regimex) Duloxetine (Cymbalta) Benztropine (Cogentin) Entacapone (Comtan) Bismuth Ergotamine and caffeine (Cafergot; subcitrate/metronidazole/tetracycline Migergot) (Pylera) Escitalopram (Lexapro) Bismuth subsalicylate (Pepto-Bismol) Fluoxetine (Prozac; Sarafem) -

A Brief Overview of Psychotropic Medication Use for Persons with Intellectual Disabilities
A BRIEF OVERVIEW OF PSYCHOTROPIC MEDICATION USE FOR PERSONS WITH INTELLECTUAL DISABILITIES INTRODUCTION Individuals with intellectual disabilities are not uncommonly prescribed psychotropic medications. Too often, historically, such agents have been used to try to improve behavioral control without adequate understanding of the antecedents, purpose, and reinforcement of the problematic behavior. While an individual with an intellectual disability may experience a depressive, anxiety, or psychotic disorder in the more typical sense some individuals experience a pattern of anxiety/alarm/arousal leading to affective dysregulation and impulsive behavior. The anxiety can be stimulated by environmental change, physical discomfort, cues related to past trauma, overstimulation, boredom, confusion, or other unpleasant states. Addressing what is causing the distress or reinforcing the behavioral response is the most important thing (though not always easy). Psychotropic medications may be useful for treating more typically presenting psychiatric illnesses as well as being part of more comprehensive plans to attenuate risk behaviors. An individual with intellectual disabilities who seems sad, is withdrawn, shows low energy and lack of interest, is eating or sleeping more or less, or may be more irritable could be suffering from a depression that needs medication treatment. On the other hand an individual with intellectual disabilities who demonstrates aggression, property destruction, self-injury, or other forms of “dyscontrol” may be helped by medication aimed at blunting the anxiety/alarm and/or blocking its escalation into aggression or other dangerous behaviors. In such instances the medications are just part of an overall strategy or plan to help the individual avoid the “need” to engage in such behavior. -

CENTRAL NERVOUS SYSTEM DEPRESSANTS Opioid Pain Relievers Anxiolytics (Also Belong to Psychiatric Medication Category) • Codeine (In 222® Tablets, Tylenol® No
CENTRAL NERVOUS SYSTEM DEPRESSANTS Opioid Pain Relievers Anxiolytics (also belong to psychiatric medication category) • codeine (in 222® Tablets, Tylenol® No. 1/2/3/4, Fiorinal® C, Benzodiazepines Codeine Contin, etc.) • heroin • alprazolam (Xanax®) • hydrocodone (Hycodan®, etc.) • chlordiazepoxide (Librium®) • hydromorphone (Dilaudid®) • clonazepam (Rivotril®) • methadone • diazepam (Valium®) • morphine (MS Contin®, M-Eslon®, Kadian®, Statex®, etc.) • flurazepam (Dalmane®) • oxycodone (in Oxycocet®, Percocet®, Percodan®, OxyContin®, etc.) • lorazepam (Ativan®) • pentazocine (Talwin®) • nitrazepam (Mogadon®) • oxazepam ( Serax®) Alcohol • temazepam (Restoril®) Inhalants Barbiturates • gases (e.g. nitrous oxide, “laughing gas”, chloroform, halothane, • butalbital (in Fiorinal®) ether) • secobarbital (Seconal®) • volatile solvents (benzene, toluene, xylene, acetone, naptha and hexane) Buspirone (Buspar®) • nitrites (amyl nitrite, butyl nitrite and cyclohexyl nitrite – also known as “poppers”) Non-Benzodiazepine Hypnotics (also belong to psychiatric medication category) • chloral hydrate • zopiclone (Imovane®) Other • GHB (gamma-hydroxybutyrate) • Rohypnol (flunitrazepam) CENTRAL NERVOUS SYSTEM STIMULANTS Amphetamines Caffeine • dextroamphetamine (Dexadrine®) Methelynedioxyamphetamine (MDA) • methamphetamine (“Crystal meth”) (also has hallucinogenic actions) • methylphenidate (Biphentin®, Concerta®, Ritalin®) • mixed amphetamine salts (Adderall XR®) 3,4-Methelynedioxymethamphetamine (MDMA, Ecstasy) (also has hallucinogenic actions) Cocaine/Crack -

Georgia State Forensic Drugs
Comprehensive Forensic FT-IR Collection Library Listing – 4,286 spectra This extensive library contains materials not only of forensic interest but also for general problem solving and identification of unknown substances in industry and academia. The wide range of items include drugs, clandestine lab chemicals, explosives, paints, fabrics, dyes, polymers, inorganic compounds, pigments, adhesives, and other common materials. The library consists of 4,286 spectra that were acquired from a wide range of laboratories involved in forensic investigations. The collection includes the following classes of compounds: • Drugs of abuse, scheduled materials • Pharmaceuticals, vitamins and excipients • Clandestine lab materials and intermediates • Solvents, organic chemicals and hazardous chemicals • Accelerants • Lubricants and natural oils • Explosives, pyrotechnics, primers, powders and boosters • Herbal and plant material and fibers • Automobile paint vehicles, pigments, primers and clear coats • Textiles, natural and man-made fibers, carpet materials • Paints, coatings, varnishes, oils • Dyes and stains • Polymers, monomers, copolymers, plasticizers and rubbers • Inorganics, pigments, minerals and clays • Tape, adhesives, sealants, glues, caulks and putties • Crystal test derivatives and intermediates • Household chemicals, cleaning agents, surfactants and pesticide All spectra were measured using micro or macro Diamond ATR, thin films on salt windows or KBr pellets at 4 cm-1 spectral resolution. Comprehensive Forensic FT-IR Collection Index -

Drugs Which Can Affect Near Vision: a Useful List
Drugs Which Can Affect Near Vision: A Useful List Joanne L. Smith B.Sc., Ph.Phm.* J. Raymond Buncic, M.D., F.R.C.S.(C)t ABSTRACT This paper documents a list of drugs that cause problems with near vision, by virtue of effects on accommodation, occasionally refractive error and diplopia. It is meant as a reference aid to the clinician when confronted with problems of focusing on near objects or print. There are many drugs that have been reported to interfere with near or reading vision, producing blurring, decreased accommodation and diplopia. This paper lists the drugs that have been reported in the literature to produce symptoms which interfere with near vision. Case reports for the listed drugs vary greatly from many to few. The drugs have been divided into the following categories: those causing (A) blurring at near, (B) diplopia and (C) induced myopia. Those drugs which only rarely cause these symptoms have been omitted. From the Departments of Pharmacy* and Ophthalmologyt, The Hospital For Sick Children, Toronto, Ontario, Canada Requests for reprints should be addressed to: Dr. J. Raymond Buncic, Department of Ophthalmology, The Hospital For Sick Children, 555 University Ave., Toronto, Ontario, Canada M5G lX8 TABLE 1 DRUGS COMMONLY CAUSING DIFFICULTY WITH FOCUSING AT NEAR OR BLURRED VISION. DRUG INCIDENCE REFERENCE Antipsychotics Chlorpromazine 14-23 8 Clozapine 5 8,14 Fluphenazine 1.2-4.3 8 Haloperidol 6.8-16 8 Loxapine 12,14 Perphenazine 7.4-17.8 8 Pimozide 20 8 Risperidone 1-2%, >/= 10% 11 Thioridazine 0.6-18 8 Thiothixene 20 8 -

West Essex CCG Anticholinergic Side-Effects and Prescribing Guidance
Anticholinergic side-effects and prescribing guidance . Anticholinergic (antimuscarinic) medications: associated with increased risks of impaired cognition and falls in patients over the age of 65 years. Recent research also points to a link to mortality increasing with the number and potency of anticholinergic agents prescribed. Anticholinergic Syndrome: is a state of confusion with characteristic features related to dysfunction of the autonomic parasympathetic (cholinergic) nervous system. Symptoms classified into systemic and CNS manifestations: o Systemic (peripheral) symptoms: Blurred vision, photophobia, non-reactive mydriasis, loss of accommodation response, flushed and dry skin, dry mouth, tachycardia, hypertension and fever. Gastrointestinal and urinary motility are frequently reduced o CNS symptoms: Delirium, agitation, disorientation, and visual hallucinations. Ataxia, choreoathetosis, myoclonus and seizures may also occur without peripheral symptoms. Medication Issues: several commonly prescribed medications that may not be thought of as anticholinergic have significant anticholinergic effects, which when taken with known anticholinergic medication can increase the risk of adverse effects. Many medication groups e.g. antihistamines, tricyclic antidepressants, drugs for asthma and COPD, cold preparations, hyoscine have varying degrees of anticholinergic activity and have the potential to cause Anticholinergic Syndrome . Clinicians should be aware of the risk for chronic anticholinergic toxicity and the fact that not all the symptoms may manifest in patients and if they do suffer some symptoms they could be wrongly attributed to another diagnosis Evidence . A study of patients over 65 found that 20% of participants who scored four or more had died by the end of the two year study period compared with 7% of patients with a score of zero. -

Briefing Note on Restriction to Use of Orphenadrine
MEDICINES MANAGEMENT BRIEFING NOTE No 3 Restriction to the use of orphenadrine NOVEMBER 2013 (UPDATED MARCH 2016) In September 2013 the Drugs and Therapeutics Committee (DTC) discussed the issues around increased risk of toxicity with orphenadrine compared to other anticholinergic drugs, particularly in overdose. Current available evidence suggests that orphenadrine has the highest mortality among anticholinergics in overdose and its potential for abuse is likely to be similar to other anticholinergics. As there are other drugs with much lower toxicity, and no clear advantages of orphenadrine, routine use of orphenadrine should be discouraged. The DTC has therefore decided to restrict the use of orphenadrine to a third-line choice, in patients who are not at risk of overdose. Orphenadrine 50mg tablets have since then been discontinued (Dec 2015), however orphenadrine oral solution 50mg in 5ml sugar free remains available as a third line option. Formulary choices for management of antipsychotic induced extra-pyramidal side- effects: First choice procyclidine 5mg tablets, 2.5mg/5ml oral solution (sugar free) Second choice trihexyphenidyl 2mg tablets, 5mg tablets, 5mg/5ml oral solution (benzhexol) Third choice Orphenadrine 50mg in 5ml oral solution (sugar free) (only in those who are not at risk of overdose) Background and Evidence Mental health disorders tend to carry a substantial risk of self-poisoning and suicide. One strategy to reduce deaths from self-poisoning is to identify the drugs which are more toxic in overdose than other drugs used for the same indication. N Buckley et. al. examined the fatal toxicity of antipsychotic drugs and anticholinergic drugs for the years 1983-1992. -

Medications for Ataxia Symptoms
NATIONALFA ATAXIAQ FOUNDATION FREQUENTLY ASKED QUESTIONS ABOUT... Medications for Ataxia Symptoms DISCLAIMER: This fact sheet is designed for educational purposes Depression: SSRI’s (Selective serotonin reuptake only and is not intended to serve as medical advice. The information inhibitors), SNRI’s (Selective norepinephrine-serotonin provided here should not be used for diagnosing or treating a health reuptake inhibitors)--classes of drugs for anxiety or problem or disease. It is not a substitute for professional care. All of these depression medications may have serious side effects and should only be used under a doctor’s supervision. NAF makes no representation or promise regarding the Dizziness/Vertigo: Acetazolamide (Diamox), effectiveness of any drug listed below. 4-aminopyridine, Baclofen, Clonazepam, Flunarizine, Gabapentin (Neurontin), Meclizine, Memantine, At this time, the goals of treatment of ataxia are to Ondansetron (Zofran), Scopolamine (eg. Transderm improve the quality of life for the person with ataxia. Scop Patch for motion sickness) For certain types of ataxia, such as ataxia due to Vitamin E deficiency, specific treatment of the underlying Excessive daytime sleepiness: Modafinil (Provigil) or problem may improve the ataxia itself. But for most Armodafinil (Nuvigil) kinds of ataxia, a treatment or cure for the disease is not yet available, so the focus is on identifying symp- Erectile Dysfunction: Cialis, Levitra, Viagra toms related to or caused by the ataxia, and treating those symptoms. Fatigue: Amantadine, Atomoxetine -

Treatment of Acute Neurolepticinduced Movement
Treatment of Acute Neuroleptic-Induced Movement Disorders Margaret E. Tonda, Pharm.D., and Sally K. Guthrie, Pharm.D. Acute extrapyramidal syndromes (EPS), including dystonia, parkinsonism, and akathisia, are associated with the use of virtually all neuroleptic agents. They may be alleviated by reducing the neuroleptic dosage, switching to a lower- potency drug, or administering an adjunctive agent such as an anticholinergic, amantadine, benzodiazepine, or P-blocker. Akathisia may be only partly dispelled by anticholinergics; alternatives are P-blockers, benzodiazepines, and clonidine. In patients receiving long-term neuroleptic therapy, both the prophylactic use and the duration of treatment with concomitant anti-EPS drugs are controversial. Administration of prophylactic anti-EPS drugs should be based on the likelihood that the patient will develop EPS, as well as the risk of adverse reactions resulting from extended use of the agents in a specific patient. The decision to continue anti-EPS therapy should be reevaluated frequently, especially in elderly patients. (Pharmacotherapy 1994;14( 51543-560) OUTLINE range of extrapyramidal syndromes (EPS) was Pharmacology observed. Although neuroleptics belong to several Pathophysiology and Clinical Manifestations of Drug- different chemical classes including phenothiazines, Induced EPS thioxanthenes, butyrophenones, dihydroindolones, Pseudoparkinsonism dibenzoxazepines, and diphenylbutylpiperidones, Acute Dystonic Reactions they are all dopamine antagonists and induce EPS Akathisia to various degrees.', ' Other drugs that cause Treatment central dopamine receptor blockade, such as Anticholinergic Agents metoclopramide (an antiemetic) and amoxapine Dopaminergic Agents (an antidepressant), can also produce extra- Benzodiazepines pyramidal movement disorder^.^^ P-Adrenergic Blockers in Akathisia Antipsychotic-induced EPS fall into four Other Agents categories: parkinsonism-like movements, acute Prophylaxis dystonia, akathisia, and tardive dyskinesia. -

Parkinsons Disease
Parkinson’s Disease (PD) is a progressive neurological condition. It affects about 1:1000 MAINTENANCE AND COMPLEX STAGE: PARKINSON’S DISEASE GUIDELINES people overall but about 1:100 of the elderly and up to 1:10 in nursing home residents. The MOTOR PSYCHOLOGICAL NON-MOTOR disease affects around 120,000 people in the UK. COMPLICATIONS COMPLICATIONS COMPLICATIONS PD is progressive, disabling and distressing. The National Institute for Clinical Excellence (NICE) released guidelines for the treatment and Freezing/festination Anxiety levels Pain management of PD in June 2006. These are soon to be reviewed. The Wirral PD Planning End of dose deterioration Depression Fatigue Group have developed these guidelines to assist you in managing patients with this complex On/Off phenomenon Panic attacks Dysphagia disease. Dyskinesias Paranoia Autonomic Dysfunction i.e. DIAGNOSIS STAGE: PARKINSON’S DISEASE GUIDELINES Dystonia Psychosis Sexual dysfunction Postural Instability Dementia Bowel & Bladder problems PD Suspected Sleep Disorders Features of PD are:- • Slowness • Stiffness Referral to PDNS (new referrals should be seen Refer to • Tremor (at rest, usually starts unilaterally) Consider referral for: within 2 weeks - NICE 2006) Specialising Consultant • Loss of balance/Falls Physio and OT Speech/Language therapy Dietician Social services Regular review and Check Prescription for Antidopaminergics Financial Support monitoring of patients e.g. Prochlorperazine (Stemetil), Metoclopramide, Haloperidol and other anti-psychotics USEFUL CONTACTS PDNS (Parkinson’s