A Dissertation Entitled Development of a Pharmacological Screen for M5
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
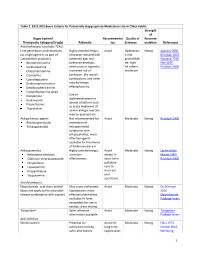
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Cell Line Descriptions Geneblazer® M1-NFAT
Version No.: GeneBLAzer ® Validation Packet Page 1 of 5 01Sep08 Optimization of the GeneBLazer ® M1-NFAT-bla Jurkat Cell Line GeneBLAzer ® M1 NFAT-bla Jurkat Cells Catalog Numbers – K1710 Cell Line Descriptions GeneBLAzer ® M1-NFAT-bla Jurkat cells contain the human Acetylcholine (muscarinic) subtype 1 receptor (M1), (Accession # NM_000738 ) stably integrated into the CellSensor ® NFAT-bla Jurkat cell line. CellSensor ® NFAT-bla Jurkat cells (Cat. no. K1671) contain a beta-lactamase (bla ) reporter gene under control of the Nuclear Factor of Activated T-cells (NFAT) response element. M1-NFAT-bla Jurkat cells are functionally validated for Z’-factor and EC 50 concentrations of carbachol (Figure 1). In addition, GeneBLAzer ® M1-NFAT-bla CHO-K1 cells have been tested for assay performance under variable conditions. Target Description Muscarinic acetylcholine receptors are members of the G protein-coupled receptor (GPCR) superfamily. Muscarinic receptors are widely distributed and mediate the actions of acetylcholine in both the CNS and peripheral tissues. Five muscarinic receptor subtypes have been identified and are referred to as M 1-M5 (1-5). The five genes that encode the muscarinic receptors all belong to the rhodopsin-line family (Family A) and share strong sequence homology but have unique regions located at the amino terminus (extracellular) and in the third intracellular loop. The M 1, M3, and M 5 receptor subtypes couple through the G q/11 class of G-proteins and activate the phopholipase C pathway. Activation of this pathway in turn leads to increases in free intracellular calcium levels as inositol triphosphate mediates release of calcium from the endoplasmic reticulum. -

The In¯Uence of Medication on Erectile Function
International Journal of Impotence Research (1997) 9, 17±26 ß 1997 Stockton Press All rights reserved 0955-9930/97 $12.00 The in¯uence of medication on erectile function W Meinhardt1, RF Kropman2, P Vermeij3, AAB Lycklama aÁ Nijeholt4 and J Zwartendijk4 1Department of Urology, Netherlands Cancer Institute/Antoni van Leeuwenhoek Hospital, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands; 2Department of Urology, Leyenburg Hospital, Leyweg 275, 2545 CH The Hague, The Netherlands; 3Pharmacy; and 4Department of Urology, Leiden University Hospital, P.O. Box 9600, 2300 RC Leiden, The Netherlands Keywords: impotence; side-effect; antipsychotic; antihypertensive; physiology; erectile function Introduction stopped their antihypertensive treatment over a ®ve year period, because of side-effects on sexual function.5 In the drug registration procedures sexual Several physiological mechanisms are involved in function is not a major issue. This means that erectile function. A negative in¯uence of prescrip- knowledge of the problem is mainly dependent on tion-drugs on these mechanisms will not always case reports and the lists from side effect registries.6±8 come to the attention of the clinician, whereas a Another way of looking at the problem is drug causing priapism will rarely escape the atten- combining available data on mechanisms of action tion. of drugs with the knowledge of the physiological When erectile function is in¯uenced in a negative mechanisms involved in erectile function. The way compensation may occur. For example, age- advantage of this approach is that remedies may related penile sensory disorders may be compen- evolve from it. sated for by extra stimulation.1 Diminished in¯ux of In this paper we will discuss the subject in the blood will lead to a slower onset of the erection, but following order: may be accepted. -

(12) Patent Application Publication (10) Pub. No.: US 2006/0110449 A1 Lorber Et Al
US 200601 10449A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0110449 A1 LOrber et al. (43) Pub. Date: May 25, 2006 (54) PHARMACEUTICAL COMPOSITION Publication Classification (76) Inventors: Richard R. Lorber, Scotch Plains, NJ (US); Heribert W. Staudinger, Union, (51) Int. Cl. NJ (US); Robert E. Ward, Summit, NJ A6II 3/473 (2006.01) (US) A6II 3L/24 (2006.01) Correspondence Address: A6IR 9/20 (2006.01) SCHERING-PLOUGH CORPORATION (52) U.S. Cl. ........................... 424/464; 514/290: 514/540 PATENT DEPARTMENT (K-6-1, 1990) 2000 GALLOPNG HILL ROAD KENILWORTH, NJ 07033-0530 (US) (57) ABSTRACT (21) Appl. No.: 11/257,348 (22) Filed: Oct. 24, 2005 The present invention relates to formulations useful for Related U.S. Application Data treating respiratory disorders associated with the production of mucus glycoprotein, skin disorders, and allergic conjunc (60) Provisional application No. 60/622,507, filed on Oct. tivitis while substantially reducing adverse effects associ 27, 2004. Provisional application No. 60/621,783, ated with the administration of non-selective anti-cholin filed on Oct. 25, 2004. ergic agents and methods of use thereof. US 2006/01 10449 A1 May 25, 2006 PHARMACEUTICAL COMPOSITION example, Weinstein and Weinstein (U.S. Pat. No. 6,086,914) describe methods of treating allergic rhinitis using an anti CROSS REFERENCE TO RELATED cholinergic agent with a limited capacity to pass across lipid APPLICATION membranes, such as the blood-brain barrier, in combination with an antihistamine that is limited in both sedating and 0001. This application claims benefit of priority to U.S. -

Drugs to Avoid in Patients with Dementia
Detail-Document #240510 -This Detail-Document accompanies the related article published in- PHARMACIST’S LETTER / PRESCRIBER’S LETTER May 2008 ~ Volume 24 ~ Number 240510 Drugs To Avoid in Patients with Dementia Elderly people with dementia often tolerate drugs less favorably than healthy older adults. Reasons include increased sensitivity to certain side effects, difficulty with adhering to drug regimens, and decreased ability to recognize and report adverse events. Elderly adults with dementia are also more prone than healthy older persons to develop drug-induced cognitive impairment.1 Medications with strong anticholinergic (AC) side effects, such as sedating antihistamines, are well- known for causing acute cognitive impairment in people with dementia.1-3 Anticholinergic-like effects, such as urinary retention and dry mouth, have also been identified in drugs not typically associated with major AC side effects (e.g., narcotics, benzodiazepines).3 These drugs are also important causes of acute confusional states. Factors that may determine whether a patient will develop cognitive impairment when exposed to ACs include: 1) total AC load (determined by number of AC drugs and dose of agents utilized), 2) baseline cognitive function, and 3) individual patient pharmacodynamic and pharmacokinetic features (e.g., renal/hepatic function).1 Evidence suggests that impairment of cholinergic transmission plays a key role in the development of Alzheimer’s dementia. Thus, the development of the cholinesterase inhibitors (CIs). When used appropriately, the CIs (donepezil [Aricept], rivastigmine [Exelon], and galantamine [Razadyne, Reminyl in Canada]) may slow the decline of cognitive and functional impairment in people with dementia. In order to achieve maximum therapeutic effect, they ideally should not be used in combination with ACs, agents known to have an opposing mechanism of action.1,2 Roe et al studied AC use in 836 elderly patients.1 Use of ACs was found to be greater in patients with probable dementia than healthy older adults (33% vs. -

In Vivo Olanzapine Occupancy of Muscarinic Acetylcholine Receptors in Patients with Schizophrenia Thomas J
In Vivo Olanzapine Occupancy of Muscarinic Acetylcholine Receptors in Patients with Schizophrenia Thomas J. Raedler, M.D., Michael B. Knable, D.O., Douglas W. Jones, Ph.D., Todd Lafargue, M.D., Richard A. Urbina, B.A., Michael F. Egan, M.D., David Pickar, M.D. , and Daniel R. Weinberger, M.D. Olanzapine is an atypical antipsychotic with potent than low-dose in the same regions. Muscarinic occupancy ϭ antimuscarinic properties in vitro (Ki 2–25 nM). We by olanzapine ranged from 13% to 57% at 5 mg/dy and studied in vivo muscarinic receptor occupancy by 26% to 79% at 20 mg/dy with an anatomical pattern olanzapine at both low dose (5 mg/dy) and high dose (20 indicating M2 subtype selectivity. The [I-123]IQNB data mg/dy) in several regions of cortex, striatum, thalamus and indicate that olanzapine is a potent and subtype-selective pons by analyzing [I-123]IQNB SPECT images of seven muscarinic antagonist in vivo, perhaps explaining its low schizophrenia patients. Both low-dose and high-dose extrapyramidal side effect profile and low incidence of olanzapine studies revealed significantly lower anticholinergic side effects. [Neuropsychopharmacology [I-123]IQNB binding than that of drug-free schizophrenia 23:56–68, 2000] Published by Elsevier Science Inc. on patients (N ϭ 12) in all regions except striatum. behalf of the American College of Neuropsychopharmacology [I-123]IQNB binding was significantly lower at high-dose KEY WORDS: Muscarinic receptor; Olanzapine; IQNB; cologically (Watling et al. 1995) and correspond to SPECT; Schizophrenia; Antipsychotic genes (respectively, m1–m5) that have been cloned (Bonner et al. -

Hypersalivation in Children and Adults
Pharmacological Management of Hypersalivation in Children and Adults Scope: Adult patients with Parkinson’s disease, children with neurodisability, cerebral palsy, long-term ventilation with drooling, and drug-induced hypersalivation. ASSESSMENT OF SEVERITY/RESPONSE TO TREATMENT: Severity of drooling can be assessed subjectively via discussion with patients and their carers/parents and by observation. Amount of drooling can be quantified by measuring the number of bibs required per day and this can also be graded using the Thomas-Stonell and Greenberg scale: • 1 = Dry (no drooling) • 2 = Mild (moist lips) • 3 = Moderate (wet lips and chin) • 4 = Severe (damp clothing) CONSIDERATIONS FOR PRESCRIBING/TITRATION No evidence to support the use of one particular treatment over another. Drug choice is to be determined by individual patient factors. When prescribing/titrating antimuscarinic drugs to treat hypersalivation always take account of: • Coexisting conditions (for example, history of urinary retention, constipation, glaucoma, dental issues, reflux etc.) • Use of other existing medication affecting the total antimuscarinic burden • Risk of adverse effects Titrate dose upward until the desired level of dryness, side effects or maximum dose reached. Take into account the preferences of the patients and their carers/ parents, and the age range and indication covered by the marketing authorisations (see individual summaries of product characteristics, BNF or BNFc for full prescribing information). FIRST LINE DRUG TREATMENT OPTIONS FOR ADULTS -

In Vitro Pharmacology of Clinically Used Central Nervous System-Active Drugs As Inverse H1 Receptor Agonists
0022-3565/07/3221-172–179$20.00 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 322, No. 1 Copyright © 2007 by The American Society for Pharmacology and Experimental Therapeutics 118869/3215703 JPET 322:172–179, 2007 Printed in U.S.A. In Vitro Pharmacology of Clinically Used Central Nervous System-Active Drugs as Inverse H1 Receptor Agonists R. A. Bakker,1 M. W. Nicholas,2 T. T. Smith, E. S. Burstein, U. Hacksell, H. Timmerman, R. Leurs, M. R. Brann, and D. M. Weiner Department of Medicinal Chemistry, Leiden/Amsterdam Center for Drug Research, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands (R.A.B., H.T., R.L.); ACADIA Pharmaceuticals Inc., San Diego, California (R.A.B., M.W.N., T.T.S., E.S.B., U.H., M.R.B., D.M.W.); and Departments of Pharmacology (M.R.B.), Neurosciences (D.M.W.), and Psychiatry (D.M.W.), University of California, San Diego, California Received January 2, 2007; accepted March 30, 2007 Downloaded from ABSTRACT The human histamine H1 receptor (H1R) is a prototypical G on this screen, we have reported on the identification of 8R- protein-coupled receptor and an important, well characterized lisuride as a potent stereospecific partial H1R agonist (Mol target for the development of antagonists to treat allergic con- Pharmacol 65:538–549, 2004). In contrast, herein we report on jpet.aspetjournals.org ditions. Many neuropsychiatric drugs are also known to po- a large number of varied clinical and chemical classes of drugs tently antagonize this receptor, underlying aspects of their side that are active in the central nervous system that display potent effect profiles. -

Nomination Background: Ketamine Hydrochloride (CASRN: 1867-66-9)
KETAMINE Nomination TABLE OF CONTENTS Page 1.0 BASIS OF NOMINATION 1 2.0 BACKGROUND INFORMATION 1 3.0 CHEMICAL PROPERTIES 2 3.1 Chemical Identification 2 3.2 Physico-Chemical Properties 3 3.3 Purity and Commercial Availability 4 4.0 PRODUCTION PROCESSES AND ANALYSIS 6 5.0 PRODUCTION AND IMPORT VOLUMES 7 6.0 USES 7 7.0 ENVIRONMENTAL OCCURRENCE 7 8.0 HUMAN EXPOSURE 7 9.0 REGULATORY STATUS 7 10.0 CLINICAL PHARMACOLOGY 7 11.0 TOXICOLOGICAL DATA 13 11.1 General Toxicology 13 11.2 Neurotoxicology 14 12.0 CONCLUSIONS 15 APPENDIX A 17 APPENDIX B 23 APPENDIX C 27 1 KETAMINE 1.0 BASIS OF NOMINATION Ketamine, a noncompetitive NMDA receptor blocker, has been used extensively off - label as a pediatric anesthetic for surgical procedures in infants and toddlers. Recently, Olney and coworkers have demonstrated severe widespread apoptotic degeneration throughout the rapidly developing brain of the 7-day-old rat after ketamine administration. Recent research at FDA has confirmed and extended Olney’s observations. These findings are cause for concern with respect to ketamine use in children. The issue of whether the neurotoxicity found in this animal model (rat) has scientific and regulatory relevance for the pediatric use of ketamine relies heavily upon confirmation of these findings that may be obtained from the conduct of an appropriate study in non-human primates. 2.0 BACKGROUND INFORMATION The issue of potential ketamine neurotoxicity in children surfaced as a result of FDA’s reluctance to approve an NIH pediatric clinical trial using this compound because of its documented neurotoxic effects in young rats (published in several papers over the last ten years by Olney and co-workers). -

The Anticholinergic Toxidrome
Poison HOTLINE Partnership between Iowa Health System and University of Iowa Hospitals and Clinics July 2011 The Anticholinergic Toxidrome A toxidrome is a group of symptoms associated with poisoning by a particular class of agents. One example is the opiate toxidrome, the triad of CNS depression, respiratory depression, and pinpoint pupils, and which usually responds to naloxone. The anticholinergic toxidrome is most frequently associated with overdoses of diphenhydramine, a very common OTC medication. However, many drugs and plants can produce the anticholinergic toxidrome. A partial list includes: tricyclic antidepressants (amitriptyline), older antihistamines (chlorpheniramine), Did you know …… phenothiazines (promethazine) and plants containing the anticholinergic alkaloids atropine, hyoscyamine and scopolamine (Jimson Weed). Each summer, the ISPCC receives approximately 10-20 The mnemonic used to help remember the symptoms and signs of this snake bite calls, some being toxidrome are derived from the Alice in Wonderland story: from poisonous snakes (both Blind as a Bat (mydriasis and inability to focus on near objects) local and exotic). Red as a Beet (flushed skin color) Four poisonous snakes can be Hot as Hades (elevated temperature) found in Iowa: the prairie These patients can sometimes die of agitation-induced hyperthermia. rattlesnake, the massasauga, Dry as a Bone (dry mouth and dry skin) the copperhead, and the Mad as a Hatter (hallucinations and delirium) timber rattlesnake. Each Bowel and bladder lose their tone (urinary retention and constipation) snake has specific territories Heart races on alone (tachycardia) within the state. ISPCC A patient who has ingested only an anticholinergic substance and is not specialists have access to tachycardic argues against a serious anticholinergic overdose. -

TE INI (19 ) United States (12 ) Patent Application Publication ( 10) Pub
US 20200187851A1TE INI (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No .: US 2020/0187851 A1 Offenbacher et al. (43 ) Pub . Date : Jun . 18 , 2020 ( 54 ) PERIODONTAL DISEASE STRATIFICATION (52 ) U.S. CI. AND USES THEREOF CPC A61B 5/4552 (2013.01 ) ; G16H 20/10 ( 71) Applicant: The University of North Carolina at ( 2018.01) ; A61B 5/7275 ( 2013.01) ; A61B Chapel Hill , Chapel Hill , NC (US ) 5/7264 ( 2013.01 ) ( 72 ) Inventors: Steven Offenbacher, Chapel Hill , NC (US ) ; Thiago Morelli , Durham , NC ( 57 ) ABSTRACT (US ) ; Kevin Lee Moss, Graham , NC ( US ) ; James Douglas Beck , Chapel Described herein are methods of classifying periodontal Hill , NC (US ) patients and individual teeth . For example , disclosed is a method of diagnosing periodontal disease and / or risk of ( 21) Appl. No .: 16 /713,874 tooth loss in a subject that involves classifying teeth into one of 7 classes of periodontal disease. The method can include ( 22 ) Filed : Dec. 13 , 2019 the step of performing a dental examination on a patient and Related U.S. Application Data determining a periodontal profile class ( PPC ) . The method can further include the step of determining for each tooth a ( 60 ) Provisional application No.62 / 780,675 , filed on Dec. Tooth Profile Class ( TPC ) . The PPC and TPC can be used 17 , 2018 together to generate a composite risk score for an individual, which is referred to herein as the Index of Periodontal Risk Publication Classification ( IPR ) . In some embodiments , each stage of the disclosed (51 ) Int. Cl. PPC system is characterized by unique single nucleotide A61B 5/00 ( 2006.01 ) polymorphisms (SNPs ) associated with unique pathways , G16H 20/10 ( 2006.01 ) identifying unique druggable targets for each stage . -

3 Drugs That May Cause Delirium Or Problem Behaviors CARD 03 19 12 JUSTIFIED.Pub
Drugs that May Cause Delirium or Problem Behaviors Drugs that May Cause Delirium or Problem Behaviors This reference card lists common and especially problemac drugs that may Ancholinergics—all drugs on this side of the card. May impair cognion cause delirium or contribute to problem behaviors in people with demena. and cause psychosis. Drugs available over‐the‐counter marked with * This does not always mean the drugs should not be used, and not all such drugs are listed. If a paent develops delirium or has new problem Tricyclic Andepressants Bladder Anspasmodics behaviors, a careful review of all medicaons is recommended. Amitriptyline – Elavil Darifenacin – Enablex Be especially mindful of new medicaons. Clomipramine – Anafranil Flavoxate – Urispas Desipramine – Norpramin Anconvulsants Psychiatric Oxybutynin – Ditropan Doxepin – Sinequan Solifenacin – VESIcare All can cause delirium, e.g. All psychiatric medicaons should be Imipramine – Tofranil Tolterodine – Detrol Carbamazepine – Tegretol reviewed as possible causes, as Nortriptyline – Aventyl, Pamelor Gabapenn – Neuronn effects are unpredictable. Trospium – Sanctura Anhistamines / Allergy / Leveracetam – Keppra Notable offenders include: Insomnia / Sleep Valproic acid – Depakote Benzodiazepines e.g. Cough & Cold Medicines *Diphenhydramine – Sominex, ‐Alprazolam – Xanax *Azelasne – Astepro Pain Tylenol‐PM, others ‐Clonazepam – Klonopin *Brompheniramine – Bromax, All opiates can cause delirium if dose *Doxylamine – Unisom, Medi‐Sleep ‐Lorazepam – Avan Bromfed, Lodrane is too high or