Phosphoinositide-Dependent Signaling in Cancer: a Focus on Phospholipase C Isozymes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Silencing of Phosphoinositide-Specific
ANTICANCER RESEARCH 34: 4069-4076 (2014) Silencing of Phosphoinositide-specific Phospholipase C ε Remodulates the Expression of the Phosphoinositide Signal Transduction Pathway in Human Osteosarcoma Cell Lines VINCENZA RITA LO VASCO1, MARTINA LEOPIZZI2, DANIELA STOPPOLONI3 and CARLO DELLA ROCCA2 Departments of 1Sense Organs , 2Medicine and Surgery Sciences and Biotechnologies and 3Biochemistry Sciences “A. Rossi Fanelli”, Sapienza University, Rome, Italy Abstract. Background: Ezrin, a member of the signal transduction pathway (5). The reduction of PIP2 ezrin–radixin–moesin family, is involved in the metastatic induces ezrin dissociation from the plasma membrane (6). spread of osteosarcoma. Ezrin binds phosphatydil inositol-4,5- The levels of PIP2 are regulated by the PI-specific bisphosphate (PIP2), a crucial molecule of the phospholipase C (PI-PLC) family (7), constituting thirteen phosphoinositide signal transduction pathway. PIP2 levels are enzymes divided into six sub-families on the basis of amino regulated by phosphoinositide-specific phospholipase C (PI- acid sequence, domain structure, mechanism of recruitment PLC) enzymes. PI-PLCε isoform, a well-characterized direct and tissue distribution (7-15). PI-PLCε, a direct effector of effector of rat sarcoma (RAS), is at a unique convergence RAS (14-15), might be the point of convergence for the point for the broad range of signaling pathways that promote broad range of signalling pathways that promote the RAS GTPase-mediated signalling. Materials and Methods. By RASGTPase-mediated signalling (16). using molecular biology methods and microscopic analyses, In previous studies, we suggested a relationship between we analyzed the expression of ezrin and PLC genes after PI-PLC expression and ezrin (17-18). -

Survival-Associated Metabolic Genes in Colon and Rectal Cancers
Survival-associated Metabolic Genes in Colon and Rectal Cancers Yanfen Cui ( [email protected] ) Tianjin Cancer Institute: Tianjin Tumor Hospital https://orcid.org/0000-0001-7760-7503 Baoai Han tianjin tumor hospital He Zhang tianjin tumor hospital Zhiyong Wang tianjin tumor hospital Hui Liu tianjin tumor hospital Fei Zhang tianjin tumor hospital Ruifang Niu tianjin tumor hospital Research Keywords: colon cancer, rectal cancer, prognosis, metabolism Posted Date: December 4th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-117478/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/42 Abstract Background Uncontrolled proliferation is the most prominent biological feature of tumors. To rapidly proliferate and maximize the use of available nutrients, tumor cells regulate their metabolic behavior and the expression of metabolism-related genes (MRGs). In this study, we aimed to construct prognosis models for colon and rectal cancers, using MRGs to indicate the prognoses of patients. Methods We rst acquired the gene expression proles of colon and rectal cancers from the TCGA and GEO database, and utilized univariate Cox analysis, lasso regression, and multivariable cox analysis to identify MRGs for risk models. Then GSEA and KEGG functional enrichment analysis were utilized to identify the metabolism pathway of MRGs in the risk models and analyzed these genes comprehensively using GSCALite. Results Eight genes (CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, GPX3, and GSR) in the colon cancer risk model and six genes (TDO2, PKLR, GAMT, EARS2, ACO1, and WAS) in the rectal cancer risk model were identied successfully. Multivariate Cox analysis indicated that the models predicted overall survival accurately and independently for patients with colon or rectal cancer. -

Supplemental Materials Supplemental Table 1
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2016 Supplemental Materials Supplemental Table 1. The differentially expressed proteins from rat pancreas identified by proteomics (SAP vs. SO) No. Protein name Gene name ratio P value 1 Metallothionein Mt1m 3.35 6.34E-07 2 Neutrophil antibiotic peptide NP-2 Defa 3.3 8.39E-07 3 Ilf2 protein Ilf2 3.18 1.75E-06 4 Numb isoform o/o rCG 3.12 2.73E-06 5 Lysozyme Lyz2 3.01 5.63E-06 6 Glucagon Gcg 2.89 1.17E-05 7 Serine protease HTRA1 Htra1 2.75 2.97E-05 8 Alpha 2 macroglobulin cardiac isoform (Fragment) 2.75 2.97E-05 9 Myosin IF (Predicted) Myo1f 2.65 5.53E-05 10 Neuroendocrine secretory protein 55 Gnas 2.61 7.60E-05 11 Matrix metallopeptidase 8 Mmp8 2.57 9.47E-05 12 Protein Tnks1bp1 Tnks1bp1 2.53 1.22E-04 13 Alpha-parvin Parva 2.47 1.78E-04 14 C4b-binding protein alpha chain C4bpa 2.42 2.53E-04 15 Protein KTI12 homolog Kti12 2.41 2.74E-04 16 Protein Rab11fip5 Rab11fip5 2.41 2.84E-04 17 Protein Mcpt1l3 Mcpt1l3 2.33 4.43E-04 18 Phospholipase B-like 1 Plbd1 2.33 4.76E-04 Aldehyde dehydrogenase (NAD), cytosolic 19 2.32 4.93E-04 (Fragments) 20 Protein Dpy19l2 Dpy19l2 2.3 5.68E-04 21 Regenerating islet-derived 3 alpha, isoform CRA_a Reg3a 2.27 6.74E-04 22 60S acidic ribosomal protein P1 Rplp1 2.26 7.22E-04 23 Serum albumin Alb 2.25 7.98E-04 24 Ribonuclease 4 Rnase4 2.24 8.25E-04 25 Cct-5 protein (Fragment) Cct5 2.24 8.52E-04 26 Protein S100-A9 S100a9 2.22 9.71E-04 27 Creatine kinase M-type Ckm 2.21 1.00E-03 28 Protein Larp4b Larp4b 2.18 1.25E-03 -
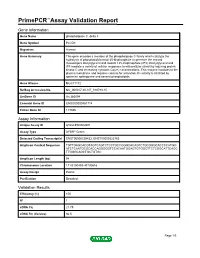
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name phospholipase C, delta 3 Gene Symbol PLCD3 Organism Human Gene Summary This gene encodes a member of the phospholipase C family which catalyze the hydrolysis of phosphatidylinositol 45-bisphosphate to generate the second messengers diacylglycerol and inositol 145-trisphosphate (IP3). Diacylglycerol and IP3 mediate a variety of cellular responses to extracellular stimuli by inducing protein kinase C and increasing cytosolic Ca(2+) concentrations. This enzyme localizes to the plasma membrane and requires calcium for activation. Its activity is inhibited by spermine sphingosine and several phospholipids. Gene Aliases MGC71172 RefSeq Accession No. NC_000017.10, NT_010783.15 UniGene ID Hs.380094 Ensembl Gene ID ENSG00000161714 Entrez Gene ID 113026 Assay Information Unique Assay ID qHsaCED0002601 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000539433, ENST00000322765 Amplicon Context Sequence TGTTGAGCACGTAGTCAGTCTCCTGCCGGGCACAGTCTGCGGGCACCCCATGG ATCTCAATGCGCACCAGGGGGTCCACAATGGAGTGTGGCTTCTCGGCATTCAGC TTGGGCAGCTGCTGTGC Amplicon Length (bp) 94 Chromosome Location 17:43190493-43190616 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 1 cDNA Cq 21.75 cDNA Tm (Celsius) 86.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 23.62 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report PLCD3, Human Amplification Plot Amplification of cDNA generated from -
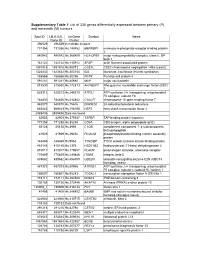
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Supplementary Table S2
1-high in cerebrotropic Gene P-value patients Definition BCHE 2.00E-04 1 Butyrylcholinesterase PLCB2 2.00E-04 -1 Phospholipase C, beta 2 SF3B1 2.00E-04 -1 Splicing factor 3b, subunit 1 BCHE 0.00022 1 Butyrylcholinesterase ZNF721 0.00028 -1 Zinc finger protein 721 GNAI1 0.00044 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 GNAI1 0.00049 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 PDE1B 0.00069 -1 Phosphodiesterase 1B, calmodulin-dependent MCOLN2 0.00085 -1 Mucolipin 2 PGCP 0.00116 1 Plasma glutamate carboxypeptidase TMX4 0.00116 1 Thioredoxin-related transmembrane protein 4 C10orf11 0.00142 1 Chromosome 10 open reading frame 11 TRIM14 0.00156 -1 Tripartite motif-containing 14 APOBEC3D 0.00173 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3D ANXA6 0.00185 -1 Annexin A6 NOS3 0.00209 -1 Nitric oxide synthase 3 SELI 0.00209 -1 Selenoprotein I NYNRIN 0.0023 -1 NYN domain and retroviral integrase containing ANKFY1 0.00253 -1 Ankyrin repeat and FYVE domain containing 1 APOBEC3F 0.00278 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3F EBI2 0.00278 -1 Epstein-Barr virus induced gene 2 ETHE1 0.00278 1 Ethylmalonic encephalopathy 1 PDE7A 0.00278 -1 Phosphodiesterase 7A HLA-DOA 0.00305 -1 Major histocompatibility complex, class II, DO alpha SOX13 0.00305 1 SRY (sex determining region Y)-box 13 ABHD2 3.34E-03 1 Abhydrolase domain containing 2 MOCS2 0.00334 1 Molybdenum cofactor synthesis 2 TTLL6 0.00365 -1 Tubulin tyrosine ligase-like family, member 6 SHANK3 0.00394 -1 SH3 and multiple ankyrin repeat domains 3 ADCY4 0.004 -1 Adenylate cyclase 4 CD3D 0.004 -1 CD3d molecule, delta (CD3-TCR complex) (CD3D), transcript variant 1, mRNA. -

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

(12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown Et Al
US 20030082511A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown et al. (43) Pub. Date: May 1, 2003 (54) IDENTIFICATION OF MODULATORY Publication Classification MOLECULES USING INDUCIBLE PROMOTERS (51) Int. Cl." ............................... C12O 1/00; C12O 1/68 (52) U.S. Cl. ..................................................... 435/4; 435/6 (76) Inventors: Steven J. Brown, San Diego, CA (US); Damien J. Dunnington, San Diego, CA (US); Imran Clark, San Diego, CA (57) ABSTRACT (US) Correspondence Address: Methods for identifying an ion channel modulator, a target David B. Waller & Associates membrane receptor modulator molecule, and other modula 5677 Oberlin Drive tory molecules are disclosed, as well as cells and vectors for Suit 214 use in those methods. A polynucleotide encoding target is San Diego, CA 92121 (US) provided in a cell under control of an inducible promoter, and candidate modulatory molecules are contacted with the (21) Appl. No.: 09/965,201 cell after induction of the promoter to ascertain whether a change in a measurable physiological parameter occurs as a (22) Filed: Sep. 25, 2001 result of the candidate modulatory molecule. Patent Application Publication May 1, 2003 Sheet 1 of 8 US 2003/0082511 A1 KCNC1 cDNA F.G. 1 Patent Application Publication May 1, 2003 Sheet 2 of 8 US 2003/0082511 A1 49 - -9 G C EH H EH N t R M h so as se W M M MP N FIG.2 Patent Application Publication May 1, 2003 Sheet 3 of 8 US 2003/0082511 A1 FG. 3 Patent Application Publication May 1, 2003 Sheet 4 of 8 US 2003/0082511 A1 KCNC1 ITREXCHO KC 150 mM KC 2000000 so 100 mM induced Uninduced Steady state O 100 200 300 400 500 600 700 Time (seconds) FIG. -

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Transcriptomic Uniqueness and Commonality of the Ion Channels and Transporters in the Four Heart Chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A
www.nature.com/scientificreports OPEN Transcriptomic uniqueness and commonality of the ion channels and transporters in the four heart chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A. Iacobas3,4* Myocardium transcriptomes of left and right atria and ventricles from four adult male C57Bl/6j mice were profled with Agilent microarrays to identify the diferences responsible for the distinct functional roles of the four heart chambers. Female mice were not investigated owing to their transcriptome dependence on the estrous cycle phase. Out of the quantifed 16,886 unigenes, 15.76% on the left side and 16.5% on the right side exhibited diferential expression between the atrium and the ventricle, while 5.8% of genes were diferently expressed between the two atria and only 1.2% between the two ventricles. The study revealed also chamber diferences in gene expression control and coordination. We analyzed ion channels and transporters, and genes within the cardiac muscle contraction, oxidative phosphorylation, glycolysis/gluconeogenesis, calcium and adrenergic signaling pathways. Interestingly, while expression of Ank2 oscillates in phase with all 27 quantifed binding partners in the left ventricle, the percentage of in-phase oscillating partners of Ank2 is 15% and 37% in the left and right atria and 74% in the right ventricle. The analysis indicated high interventricular synchrony of the ion channels expressions and the substantially lower synchrony between the two atria and between the atrium and the ventricle from the same side. Starting with crocodilians, the heart pumps the blood through the pulmonary circulation and the systemic cir- culation by the coordinated rhythmic contractions of its upper lef and right atria (LA, RA) and lower lef and right ventricles (LV, RV). -

Protein Network Analyses of Pulmonary Endothelial Cells In
www.nature.com/scientificreports OPEN Protein network analyses of pulmonary endothelial cells in chronic thromboembolic pulmonary hypertension Sarath Babu Nukala1,8,9*, Olga Tura‑Ceide3,4,5,9, Giancarlo Aldini1, Valérie F. E. D. Smolders2,3, Isabel Blanco3,4, Victor I. Peinado3,4, Manuel Castell6, Joan Albert Barber3,4, Alessandra Altomare1, Giovanna Baron1, Marina Carini1, Marta Cascante2,7,9 & Alfonsina D’Amato1,9* Chronic thromboembolic pulmonary hypertension (CTEPH) is a vascular disease characterized by the presence of organized thromboembolic material in pulmonary arteries leading to increased vascular resistance, heart failure and death. Dysfunction of endothelial cells is involved in CTEPH. The present study describes for the frst time the molecular processes underlying endothelial dysfunction in the development of the CTEPH. The advanced analytical approach and the protein network analyses of patient derived CTEPH endothelial cells allowed the quantitation of 3258 proteins. The 673 diferentially regulated proteins were associated with functional and disease protein network modules. The protein network analyses resulted in the characterization of dysregulated pathways associated with endothelial dysfunction, such as mitochondrial dysfunction, oxidative phosphorylation, sirtuin signaling, infammatory response, oxidative stress and fatty acid metabolism related pathways. In addition, the quantifcation of advanced oxidation protein products, total protein carbonyl content, and intracellular reactive oxygen species resulted increased