Small Molecule Protein–Protein Interaction Inhibitors As CNS Therapeutic Agents: Current Progress and Future Hurdles
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Silencing of Phosphoinositide-Specific
ANTICANCER RESEARCH 34: 4069-4076 (2014) Silencing of Phosphoinositide-specific Phospholipase C ε Remodulates the Expression of the Phosphoinositide Signal Transduction Pathway in Human Osteosarcoma Cell Lines VINCENZA RITA LO VASCO1, MARTINA LEOPIZZI2, DANIELA STOPPOLONI3 and CARLO DELLA ROCCA2 Departments of 1Sense Organs , 2Medicine and Surgery Sciences and Biotechnologies and 3Biochemistry Sciences “A. Rossi Fanelli”, Sapienza University, Rome, Italy Abstract. Background: Ezrin, a member of the signal transduction pathway (5). The reduction of PIP2 ezrin–radixin–moesin family, is involved in the metastatic induces ezrin dissociation from the plasma membrane (6). spread of osteosarcoma. Ezrin binds phosphatydil inositol-4,5- The levels of PIP2 are regulated by the PI-specific bisphosphate (PIP2), a crucial molecule of the phospholipase C (PI-PLC) family (7), constituting thirteen phosphoinositide signal transduction pathway. PIP2 levels are enzymes divided into six sub-families on the basis of amino regulated by phosphoinositide-specific phospholipase C (PI- acid sequence, domain structure, mechanism of recruitment PLC) enzymes. PI-PLCε isoform, a well-characterized direct and tissue distribution (7-15). PI-PLCε, a direct effector of effector of rat sarcoma (RAS), is at a unique convergence RAS (14-15), might be the point of convergence for the point for the broad range of signaling pathways that promote broad range of signalling pathways that promote the RAS GTPase-mediated signalling. Materials and Methods. By RASGTPase-mediated signalling (16). using molecular biology methods and microscopic analyses, In previous studies, we suggested a relationship between we analyzed the expression of ezrin and PLC genes after PI-PLC expression and ezrin (17-18). -

A Core Program of Gene Expression Characterizes Cancer Metastases
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 60), pp: 102161-102175 Research Paper A core program of gene expression characterizes cancer metastases Franz Hartung1, Yunguan Wang2, Bruce Aronow2 and Georg F. Weber1 1University of Cincinnati Academic Health Center, Cincinnati, OH, USA 2Computational Medicine Center, Cincinnati Children’s Hospital, Cincinnati, OH, USA Correspondence to: Georg F. Weber, email: [email protected] Keywords: metastasis; metabolism; vascularization; extracellular matrix; ion homeostasis Received: July 28, 2017 Accepted: August 31, 2017 Published: November 02, 2017 Copyright: Hartung et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT While aberrant expression or splicing of metastasis genes conveys to cancers the ability to break through tissue barriers and disseminate, the genetic basis for organ preference in metastasis formation has remained incompletely understood. Utilizing the gene expression profiles from 653 GEO datasets, we investigate whether the signatures by diverse cancers in various metastatic sites display common features. We corroborate the meta-analysis in a murine model. Metastases are generally characterized by a core program of gene expression that induces the oxidative metabolism, activates vascularization/tissue remodeling, silences extracellular matrix interactions, and alters ion homeostasis. This program distinguishes metastases from their originating primary tumors as well as from their target host tissues. Site- selectivity is accomplished through specific components that adjust to the target micro-environment. The same functional groups of gene expression programs are activated in the metastases of B16-F10 cells to various target organs. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Survival-Associated Metabolic Genes in Colon and Rectal Cancers
Survival-associated Metabolic Genes in Colon and Rectal Cancers Yanfen Cui ( [email protected] ) Tianjin Cancer Institute: Tianjin Tumor Hospital https://orcid.org/0000-0001-7760-7503 Baoai Han tianjin tumor hospital He Zhang tianjin tumor hospital Zhiyong Wang tianjin tumor hospital Hui Liu tianjin tumor hospital Fei Zhang tianjin tumor hospital Ruifang Niu tianjin tumor hospital Research Keywords: colon cancer, rectal cancer, prognosis, metabolism Posted Date: December 4th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-117478/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/42 Abstract Background Uncontrolled proliferation is the most prominent biological feature of tumors. To rapidly proliferate and maximize the use of available nutrients, tumor cells regulate their metabolic behavior and the expression of metabolism-related genes (MRGs). In this study, we aimed to construct prognosis models for colon and rectal cancers, using MRGs to indicate the prognoses of patients. Methods We rst acquired the gene expression proles of colon and rectal cancers from the TCGA and GEO database, and utilized univariate Cox analysis, lasso regression, and multivariable cox analysis to identify MRGs for risk models. Then GSEA and KEGG functional enrichment analysis were utilized to identify the metabolism pathway of MRGs in the risk models and analyzed these genes comprehensively using GSCALite. Results Eight genes (CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, GPX3, and GSR) in the colon cancer risk model and six genes (TDO2, PKLR, GAMT, EARS2, ACO1, and WAS) in the rectal cancer risk model were identied successfully. Multivariate Cox analysis indicated that the models predicted overall survival accurately and independently for patients with colon or rectal cancer. -

Ck1δ Over-Expressing Mice Display ADHD-Like Behaviors, Frontostriatal Neuronal Abnormalities and Altered Expressions of ADHD-Candidate Genes
Molecular Psychiatry (2020) 25:3322–3336 https://doi.org/10.1038/s41380-018-0233-z ARTICLE CK1δ over-expressing mice display ADHD-like behaviors, frontostriatal neuronal abnormalities and altered expressions of ADHD-candidate genes 1 1 1 2 1 1 1 Mingming Zhou ● Jodi Gresack ● Jia Cheng ● Kunihiro Uryu ● Lars Brichta ● Paul Greengard ● Marc Flajolet Received: 8 November 2017 / Revised: 4 July 2018 / Accepted: 18 July 2018 / Published online: 19 October 2018 © Springer Nature Limited 2018 Abstract The cognitive mechanisms underlying attention-deficit hyperactivity disorder (ADHD), a highly heritable disorder with an array of candidate genes and unclear genetic architecture, remain poorly understood. We previously demonstrated that mice overexpressing CK1δ (CK1δ OE) in the forebrain show hyperactivity and ADHD-like pharmacological responses to D- amphetamine. Here, we demonstrate that CK1δ OE mice exhibit impaired visual attention and a lack of D-amphetamine- induced place preference, indicating a disruption of the dopamine-dependent reward pathway. We also demonstrate the presence of abnormalities in the frontostriatal circuitry, differences in synaptic ultra-structures by electron microscopy, as 1234567890();,: 1234567890();,: well as electrophysiological perturbations of both glutamatergic and GABAergic transmission, as observed by altered frequency and amplitude of mEPSCs and mIPSCs. Furthermore, gene expression profiling by next-generation sequencing alone, or in combination with bacTRAP technology to study specifically Drd1a versus Drd2 medium spiny neurons, revealed that developmental CK1δ OE alters transcriptional homeostasis in the striatum, including specific alterations in Drd1a versus Drd2 neurons. These results led us to perform a fine molecular characterization of targeted gene networks and pathway analysis. Importantly, a large fraction of 92 genes identified by GWAS studies as associated with ADHD in humans are significantly altered in our mouse model. -

Effective Angiogenesis Requires Regulation of Phosphoinositide Signaling
Effective angiogenesis requires regulation of phosphoinositide signaling Elizabeth Michele Davies, Rajendra Gurung, Kai Qin Le, Christina Anne Mitchell* Cancer Program, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Victoria 3800, Australia *Corresponding author [email protected] Abstract Phosphoinositide signaling regulates numerous downstream effectors that mediate cellular processes which influence cell cycle progression, migration, proliferation, growth, survival, metabolism and vesicular trafficking. A prominent role for phosphoinositide 3-kinase, which generates phosphatidylinositol 3,4,5-trisphosphate, a phospholipid that activates a plethora of effectors including AKT and FOXO during embryonic and postnatal angiogenesis, has been described. In addition, phosphatidylinositol 3-phosphate signaling is required for endosomal trafficking, which contributes to vascular remodeling. This review will examine the role phosphoinositide signaling plays in the endothelium and its contribution to sprouting angiogenesis. Keywords Angiogenesis, Phosphoinositide, PI3K, PTEN 1. Introduction Establishment of a functional vasculature is essential for embryonic development, and also for the maintenance of tissue regeneration, inflammatory responses and wound healing in the adult. Vascular development is characterized by two distinct physiological processes, vasculogenesis and angiogenesis. Vasculogenesis is predominantly limited to embryonic development, whereby differentiation -
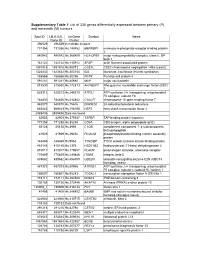
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Supplementary Table S2
1-high in cerebrotropic Gene P-value patients Definition BCHE 2.00E-04 1 Butyrylcholinesterase PLCB2 2.00E-04 -1 Phospholipase C, beta 2 SF3B1 2.00E-04 -1 Splicing factor 3b, subunit 1 BCHE 0.00022 1 Butyrylcholinesterase ZNF721 0.00028 -1 Zinc finger protein 721 GNAI1 0.00044 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 GNAI1 0.00049 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 PDE1B 0.00069 -1 Phosphodiesterase 1B, calmodulin-dependent MCOLN2 0.00085 -1 Mucolipin 2 PGCP 0.00116 1 Plasma glutamate carboxypeptidase TMX4 0.00116 1 Thioredoxin-related transmembrane protein 4 C10orf11 0.00142 1 Chromosome 10 open reading frame 11 TRIM14 0.00156 -1 Tripartite motif-containing 14 APOBEC3D 0.00173 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3D ANXA6 0.00185 -1 Annexin A6 NOS3 0.00209 -1 Nitric oxide synthase 3 SELI 0.00209 -1 Selenoprotein I NYNRIN 0.0023 -1 NYN domain and retroviral integrase containing ANKFY1 0.00253 -1 Ankyrin repeat and FYVE domain containing 1 APOBEC3F 0.00278 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3F EBI2 0.00278 -1 Epstein-Barr virus induced gene 2 ETHE1 0.00278 1 Ethylmalonic encephalopathy 1 PDE7A 0.00278 -1 Phosphodiesterase 7A HLA-DOA 0.00305 -1 Major histocompatibility complex, class II, DO alpha SOX13 0.00305 1 SRY (sex determining region Y)-box 13 ABHD2 3.34E-03 1 Abhydrolase domain containing 2 MOCS2 0.00334 1 Molybdenum cofactor synthesis 2 TTLL6 0.00365 -1 Tubulin tyrosine ligase-like family, member 6 SHANK3 0.00394 -1 SH3 and multiple ankyrin repeat domains 3 ADCY4 0.004 -1 Adenylate cyclase 4 CD3D 0.004 -1 CD3d molecule, delta (CD3-TCR complex) (CD3D), transcript variant 1, mRNA. -

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Reverse Pathway Genetic Approach Identifies Epistasis in Autism Spectrum Disorders
UCSF UC San Francisco Previously Published Works Title Reverse Pathway Genetic Approach Identifies Epistasis in Autism Spectrum Disorders. Permalink https://escholarship.org/uc/item/7mr5n558 Journal PLoS genetics, 13(1) ISSN 1553-7390 Authors Mitra, Ileena Lavillaureix, Alinoë Yeh, Erika et al. Publication Date 2017-01-11 DOI 10.1371/journal.pgen.1006516 Peer reviewed eScholarship.org Powered by the California Digital Library University of California RESEARCH ARTICLE Reverse Pathway Genetic Approach Identifies Epistasis in Autism Spectrum Disorders Ileena Mitra1,2☯, AlinoeÈ Lavillaureix1,2,3☯, Erika Yeh1,2, Michela Traglia1,2, Kathryn Tsang1,2, Carrie E. Bearden4,5, Katherine A. Rauen2,6¤, Lauren A. Weiss1,2* 1 Department of Psychiatry, University of California San Francisco, San Francisco, California, United States of America, 2 Institute for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 3 Universite Paris Descartes, Sorbonne Paris CiteÂ, Faculty of Medicine, Paris, France, 4 Department of Psychiatry and Biobehavioral Sciences, Semel Institute for Neuroscience and Human Behavior, University of California Los Angeles, Los Angeles, California, United States of America, a1111111111 5 Department of Psychology, University of California Los Angeles, Los Angeles, California, United States of a1111111111 America, 6 Department of Pediatrics, School of Medicine, University of California San Francisco, San a1111111111 Francisco, California, United States of America a1111111111 a1111111111 ☯ These authors contributed equally to this work. ¤ Current Address: Division of Genomic Medicine, Department of Pediatrics, UC Davis MIND Institute, University of California at Davis, Sacramento, California * [email protected] OPEN ACCESS Abstract Citation: Mitra I, Lavillaureix A, Yeh E, Traglia M, Tsang K, Bearden CE, et al. -

Signal Transduction in L-DOPA-Induced Dyskinesia: from Receptor Sensitization to Abnormal… Et Al
Journal of Neural Transmission https://doi.org/10.1007/s00702-018-1847-7 NEUROLOGY AND PRECLINICAL NEUROLOGICAL STUDIES - REVIEW ARTICLE Signal transduction in L‑DOPA‑induced dyskinesia: from receptor sensitization to abnormal gene expression Giada Spigolon1 · Gilberto Fisone1 Received: 1 November 2017 / Accepted: 23 January 2018 © The Author(s) 2018. This article is an open access publication Abstract A large number of signaling abnormalities have been implicated in the emergence and expression of L-DOPA-induced dyskinesia (LID). The primary cause for many of these changes is the development of sensitization at dopamine receptors located on striatal projection neurons (SPN). This initial priming, which is particularly evident at the level of dopamine D1 receptors (D1R), can be viewed as a homeostatic response to dopamine depletion and is further exacerbated by chronic administration of L-DOPA, through a variety of mechanisms afecting various components of the G-protein-coupled receptor machinery. Sensitization of dopamine receptors in combination with pulsatile administration of L-DOPA leads to intermit- tent and coordinated hyperactivation of signal transduction cascades, ultimately resulting in long-term modifcations of gene expression and protein synthesis. A detailed mapping of these pathological changes and of their involvement in LID has been produced during the last decade. According to this emerging picture, activation of sensitized D1R results in the stimulation of cAMP-dependent protein kinase and of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa. This, in turn, activates the extracellular signal-regulated kinases 1 and 2 (ERK), leading to chromatin remodeling and aberrant gene transcription. Dysregulated ERK results also in the stimulation of the mammalian target of rapamycin complex 1, which promotes protein synthesis.