Secondary Metabolism Drives Ecological Breadth in the Xylariaceae 2 3 Mario E.E
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biscogniauxia Granmoi (Xylariaceae) in Europe
©Österreichische Mykologische Gesellschaft, Austria, download unter www.biologiezentrum.at Osten. Z.Pilzk 8(1999) 139 Biscogniauxia granmoi (Xylariaceae) in Europe THOMAS L£SS0E CHRISTIAN SCHEUER Botanical Institute, Copenhagen University Institut fiir Botanik der Karl-Franzens-Universitat Oster Farimagsgade 2D Holteigasse 6 DK-1353 Copenhagen K, Denmark A-8010 Graz, Austria e-mail: [email protected] e-mail: [email protected] ALFRED GRANMO Trornso Museum, University of Tromse N-9037 Tromso, Norway e-mail: [email protected] Received 5. 7. 1999 Key words: Xylariaceae, Biscogniauxia. - Taxonomy, distribution. - Fungi of Europe, Asia. Abstract: Biscogniauxia granmoi, growing on Prunus padus (incl. var. pubescens = Padus asiatica) is reported from Europe and Asia, with material from Austria, Latvia, Norway, Poland, and Far Eastern Russia. It is compared with B. nummulana s. str., B. capnodes and B. simphcior. The taxon was included in the recent revision of Biscogniauxia by JU & al. {1998, Mycotaxon 66: 50) under the name "B. pruni GRANMO, L/ESS0E & SCHEUER" nom. prov. Zusammenfassung: Biscogniauxia granmoi, die bisher ausschließlich auf Prunus padus (inkl. var pubescens = Padus asiatica) gefunden wurde, wird aufgrund von Aufsammlungen aus Europa und Asien vorgestellt. Die bisherigen Belege stammen aus Österreich, Litauen, Norwegen, Polen und dem femöstlichen Teil Rußlands. Die Unterschiede zu B. nummulana s. Str., B. capnodes und H simphaor werden diskutiert. Dieses Taxon wurde unter dem Namen "B. pruni Granmo, l.aessoe & Scheuer" nom. prov. schon von JU & al. (1998, Mycotaxon 66: 50) in ihre Revision der Gattung Biscogniauxia aufge- nommen. The genus Biscogniauxia KUNTZE (Xylariaceae) was resurrected and amended by POUZAR (1979, 1986) for a group of Xylariaceae with applanate dark stromata that MILLER (1961) treated in Hypoxylon BULL., and for a group of species with thick, discoid stromata formerly placed in Nummularia TUL. -
Oakland Sheriff's Dept Fee Schedule 02.09
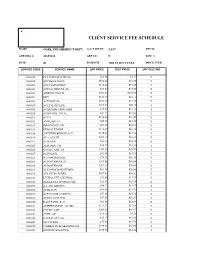
CLIENT SERVICE FEE SCHEDULE NAME: OAKLAND SHERIFF'S DEPT ACCT SETUP: 2/1/87 REP ID: GRP BILL #: 22857610 GRP CD: N DISC %: FS ID: 01 FS DESCR: 2003 CLIENT FEES PRICE TYPE: SERVICE CODE SERVICE NAME UPF PRICE TEST PRICE UPF DISC IND 0000018 PLT SODIUM CITRATE $12.86 $4.37 Y 0000022 APC RESISTANCE $101.62 $34.55 Y 0000201 ACETAMINOPHEN $110.24 $37.48 Y 0000202 ACETALDEHYDE (B) $73.00 $73.00 N 0000203 ARSENIC,TISSUE $302.00 $302.00 N 0000204 DHT $182.70 $62.12 Y 0000205 ACETONE (B) $102.20 $34.75 Y 0000206 ACETYLCHOLINE $165.00 $56.10 Y 0000208 ACID PHOS, PROS, IMM $35.60 $12.10 Y 0000210 ACID PHOS, TOTAL $37.22 $12.65 Y 0000211 ACTH $110.00 $37.40 Y 0000212 AMYLASE (U) $45.45 $15.45 Y 0000213 IMMUNOFIX, UR $67.39 $22.91 Y 0000214 ETHOSUXIMIDE $112.07 $38.10 Y 0000216 ANTITHROMBIN III ACT $170.46 $57.96 Y 0000219 ALA, QUANT $107.10 $36.41 Y 0000223 ALBUMIN $22.88 $22.88 N 0000224 ALBUMIN, CSF $36.25 $12.33 Y 0000225 CYCLIC AMP, UR $205.80 $69.97 Y 0000227 ALDOLASE $32.08 $10.91 Y 0000228 A-2-MACROGLOB. $78.75 $26.78 Y 0000229 ALDOSTERONE (U) $251.58 $85.54 Y 0000230 ALDOSTERONE $207.10 $70.41 Y 0000231 ALK PHOS ISOENZYMES $61.34 $20.86 Y 0000232 AFP, FLUID W/RFX $107.40 $36.52 Y 0000233 LEUKOCYTE ALK PHOS $34.60 $11.76 Y 0000234 ALKALINE PHOSPHATASE $22.88 $22.88 N 0000235 A-1-ANTITRYPSN $40.22 $13.67 Y 0000236 AMIKACIN $99.91 $33.97 Y 0000237 AFP,TUMOR (CHIRON) $53.86 $18.31 Y 0000238 AMINO ACID SCR $87.15 $29.63 Y 0000240 RAST PANEL #103 $62.00 $62.00 N 0000241 AMPHETAMINE GC/MS $111.70 $37.98 Y 0000242 CYCLIC AMP $205.80 $69.97 Y 0000243 AMYLASE $16.42 $5.58 Y 0000248 HACKBERRY IGE $35.40 $35.40 N 0000249 ANA W/RFX $55.00 $18.70 Y 0000250 *AMIKACIN,PEAK&TROUGH $199.83 $67.94 Y 0000251 ANDROSTENEDIONE $180.65 $61.42 Y 0000252 ANTI-DIUR. -

Mycosphere Notes 225–274: Types and Other Specimens of Some Genera of Ascomycota
Mycosphere 9(4): 647–754 (2018) www.mycosphere.org ISSN 2077 7019 Article Doi 10.5943/mycosphere/9/4/3 Copyright © Guizhou Academy of Agricultural Sciences Mycosphere Notes 225–274: types and other specimens of some genera of Ascomycota Doilom M1,2,3, Hyde KD2,3,6, Phookamsak R1,2,3, Dai DQ4,, Tang LZ4,14, Hongsanan S5, Chomnunti P6, Boonmee S6, Dayarathne MC6, Li WJ6, Thambugala KM6, Perera RH 6, Daranagama DA6,13, Norphanphoun C6, Konta S6, Dong W6,7, Ertz D8,9, Phillips AJL10, McKenzie EHC11, Vinit K6,7, Ariyawansa HA12, Jones EBG7, Mortimer PE2, Xu JC2,3, Promputtha I1 1 Department of Biology, Faculty of Science, Chiang Mai University, Chiang Mai 50200, Thailand 2 Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, 132 Lanhei Road, Kunming 650201, China 3 World Agro Forestry Centre, East and Central Asia, 132 Lanhei Road, Kunming 650201, Yunnan Province, People’s Republic of China 4 Center for Yunnan Plateau Biological Resources Protection and Utilization, College of Biological Resource and Food Engineering, Qujing Normal University, Qujing, Yunnan 655011, China 5 Shenzhen Key Laboratory of Microbial Genetic Engineering, College of Life Sciences and Oceanography, Shenzhen University, Shenzhen 518060, China 6 Center of Excellence in Fungal Research, Mae Fah Luang University, Chiang Rai 57100, Thailand 7 Department of Entomology and Plant Pathology, Faculty of Agriculture, Chiang Mai University, Chiang Mai 50200, Thailand 8 Department Research (BT), Botanic Garden Meise, Nieuwelaan 38, BE-1860 Meise, Belgium 9 Direction Générale de l'Enseignement non obligatoire et de la Recherche scientifique, Fédération Wallonie-Bruxelles, Rue A. -

Endophytic Fungi: Treasure for Anti-Cancerous Compounds
International Journal of Pharmacy and Pharmaceutical Sciences ISSN- 0975-1491 Vol 8, Issue 8, 2016 Review Article ENDOPHYTIC FUNGI: TREASURE FOR ANTI-CANCEROUS COMPOUNDS ANAND DILIP FIRODIYAa*, RAJESH KUMAR TENGURIAb aCSRD, Peoples University, Bhopal 462037, Madhya Pradesh, India, bDepartment of Botany, Govt. PG College, Rajgarh 496551, Madhya Pradesh, India Email: [email protected] Received: 22 Apr 2016 Revised and Accepted: 20 June 2016 ABSTRACT Endophytic fungi that live asymptomatically inside the plant tissues have novel bioactive metabolites exhibiting a variety of biological activities, especially against cancer. This review highlights the research progress on the production of anticancer compounds by endophytic fungi from 1990- 2015. Anticancer activity is generally associated with the cytotoxicity of the compounds present in the endophytic fungi. The ubiquitous nature of endophytic fungi synthesise diverse chemicals with promising anticancer activity from either their original host or related species. Modification in fermentation parameters and genetic insight of endophytes may produce novel anti-cancerous compounds. Keywords: Cancer, Medicinal plants, Secondary metabolites © 2016 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/) INTRODUCTION endophytic fungi detectable by high-performance liquid chromate- graphy, nuclear magnetic resonance, mass spectrophotometer and The interest in the biogenic medicines has revived throughout the X-ray crystallography and its cytotoxicity of the bioactive world, as the increase in awareness of the health hazards and compounds against cancer cell lines. The compounds with potential toxicity associated with the random use of synthetic drugs and application were also considered in the selection of antitumor antibiotics [1]. -

Sayı Tam Dosyası
']FHhQLYHUVLWHVL2UPDQFÕOÕN'HUJLVL&LOW166D\Õ2 )DNOWH$GÕQD6DKLEL : 3URI'U+DOGXQ0h'(55ø62ö/8 %Dú(GLW|U : 'Ro'U(QJLQ(52ö/8 Editör Kurulu Alan Editörleri Prof. Dr. Oktay YILDIZ 3URI'U'HU\D(ù(1 Prof. Dr. Kermit CROMAC Jr. (Oregon State University) Prof. Dr. Rimvydas VASAITIS (Swedish University of Agricultural Sciences) 3URI'U-LĜt5(0(â &]HFK8QLYHUVLW\RI/LIH6FLHQFHV3UDJXH Prof. Dr. Marc J. LINIT (University of Missouri) 3URI'U=HNL'(0ø5 Prof. Dr. (PUDKdød(. Prof. 'U'U'HU\D6(9ø0.25.87 Prof. 'U$\ELNH$\IHU.$5$'$ö Doç'U0.ÕYDQo$. Doç'U7DUÕN*('ø. Doç. Dr. Akif KETEN Doç. Dr. Ali Kemal ÖZBAYRAM 'UgJUh3ÕQDU.g</h 'UgJUh'U+DVDQg='(0ø5 Dr. Ögr. Ü. Dr. Hüseyin AMBARLI Dr. gJUh'UøGULV'85862< 'UgJUh'U%LODOd(7ø1 Teknik Editörler $Uú*|U6HUWDo.$<$ $Uú*|U0XKDPPHWdø/ $Uú*|U'UdD÷ODU$.d$< $Uú*|U'U7DUÕNdø7*(= Dr. Ögr. Ü. Ömer ÖZYÜREK $Uú*|U1XUD\g=7h5. $Uú*|U<ÕOGÕ]%$+d(&ø $Uú*|UAbdullah Hüseyin DÖNMEZ Dil Editörleri gJU*|U'UøVPDLO.2d Ögr. Gör. Dr. Zennure UÇAR zĂnjŦƔŵĂĚƌĞƐŝ ŽƌƌĞƐƉŽŶĚŝŶŐĚĚƌĞƐƐ Düzce Üniversitesi Duzce University Orman Fakültesi Faculty of Forestry ϴϭϲϮϬ<ŽŶƵƌĂůƉzĞƌůĞƔŬĞƐŝͬƺnjĐĞ-dmZ<7z ϴϭϲϮϬ<ŽŶƵƌĂůƉĂŵƉƵƐͬƺnjĐĞ-dhZ<z 'HUJL\ÕOGDLNLVD\ÕRODUDN\D\ÕQODQÕU 7KLVMRXUQDOLVSXEOLVKHGVHPLDQQXDOO\ http://www.duzce.edu.tr/of/ DGUHVLQGHQGHUJL\HLOLúNLQELOJLOHUHYHPDNDOH|]HWOHULQHXODúÕODELOLU (Instructions to Authors" and "Abstracts" can be found at this address). ødø1'(.ø/(5 +X]XUHYL%DKoHOHULQLQ<Dú'RVWX7DVDUÕP$oÕVÕQGDQøQFHOHQPHVLAntalya-7UNL\HgUQH÷L«««««1 Tahsin YILMAZ, Bensu YÜCE .HQWVHO5HNUHDV\RQHO$ODQODUGDNL%LWNL9DUOÕ÷Õ5L]HgUQH÷L«««««««««««««««««16 Ömer Lütfü ÇORBACI, *|NKDQ$%$<7UNHU2ö8=7h5.0HUYHhd2. <Õ÷ÕOFD ']FH %DON|\ %DO2UPDQÕ)ORUDVÕ««««««««««««««««««««««««45 (OLI$\úH<,/',5,01HYDO*h1(ùg=.$11XUJO.$5/,2ö/8.,/,d Assessment of Basic Green Infrastructure Components as Part of Landscape Structure for Siirt……...70 Huriye Simten SÜTÜNÇ, Ömer Lütfü ÇORBACI Cephalaria duzceënsis N. -

Genetic and Pathogenic Differences Between Microdochium Nivale and Microdochium Majus
Genetic and Pathogenic Differences Between Microdochium nivale and Microdochium majus by Linda Elizabeth Jewell A Thesis Presented to The University of Guelph In partial fulfilment of requirements for the degree of Doctor of Philosophy in Environmental Science Guelph, Ontario, Canada © Linda Elizabeth Jewell, December, 2013 ABSTRACT GENETIC AND PATHOGENIC DIFFERENCES BETWEEN MICRODOCHIUM NIVALE AND MICRODOCHIUM MAJUS Linda Elizabeth Jewell Advisor: University of Guelph, 2013 Professor Tom Hsiang Microdochium nivale and M. majus are fungal plant pathogens that cause cool-temperature diseases on grasses and cereals. Nucleotide sequences of four genetic regions were compared between isolates of M. nivale and M. majus from Triticum aestivum (wheat) collected in North America and Europe and for isolates of M. nivale from turfgrasses from both continents. Draft genome sequences were assembled for two isolates of M. majus and two of M. nivale from wheat and one from turfgrass. Dendograms constructed from these data resolved isolates of M. majus into separate clades by geographic origin. Among M. nivale, isolates were instead resolved by host plant species. Amplification of repetitive regions of DNA from M. nivale isolates collected from two proximate locations across three years grouped isolates by year, rather than by location. The mating-type (MAT1) and associated flanking genes of Microdochium were identified using the genome sequencing data to investigate the potential for these pathogens to produce ascospores. In all of the Microdochium genomes, and in all isolates assessed by PCR, only the MAT1-2-1 gene was identified. However, unpaired, single-conidium-derived colonies of M. majus produced fertile perithecia in the lab. -

Phylogenetic Assignment of the Fungicolous Hypoxylon Invadens (Ascomycota, Xylariales) and Investigation of Its Secondary Metabolites
microorganisms Article Phylogenetic Assignment of the Fungicolous Hypoxylon invadens (Ascomycota, Xylariales) and Investigation of its Secondary Metabolites Kevin Becker 1,2 , Christopher Lambert 1,2,3 , Jörg Wieschhaus 1 and Marc Stadler 1,2,* 1 Department of Microbial Drugs, Helmholtz Centre for Infection Research GmbH (HZI), Inhoffenstraße 7, 38124 Braunschweig, Germany; [email protected] (K.B.); [email protected] (C.L.); [email protected] (J.W.) 2 German Centre for Infection Research Association (DZIF), Partner site Hannover-Braunschweig, Inhoffenstraße 7, 38124 Braunschweig, Germany 3 Department for Molecular Cell Biology, Helmholtz Centre for Infection Research GmbH (HZI) Inhoffenstraße 7, 38124 Braunschweig, Germany * Correspondence: [email protected]; Tel.: +49-531-6181-4240; Fax: +49-531-6181-9499 Received: 23 July 2020; Accepted: 8 September 2020; Published: 11 September 2020 Abstract: The ascomycete Hypoxylon invadens was described in 2014 as a fungicolous species growing on a member of its own genus, H. fragiforme, which is considered a rare lifestyle in the Hypoxylaceae. This renders H. invadens an interesting target in our efforts to find new bioactive secondary metabolites from members of the Xylariales. So far, only volatile organic compounds have been reported from H. invadens, but no investigation of non-volatile compounds had been conducted. Furthermore, a phylogenetic assignment following recent trends in fungal taxonomy via a multiple sequence alignment seemed practical. A culture of H. invadens was thus subjected to submerged cultivation to investigate the produced secondary metabolites, followed by isolation via preparative chromatography and subsequent structure elucidation by means of nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HR-MS). -

And Mushroom-Associated Alkaloids from Two Behavior Modifying
bioRxiv preprint doi: https://doi.org/10.1101/375105; this version posted December 18, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Psychoactive plant- and mushroom-associated alkaloids from two behavior modifying 2 cicada pathogens 3 4 Greg R. Boyce1, Emile Gluck-Thaler2, Jason C. Slot2, Jason E. Stajich3, William J. Davis4, Tim 5 Y. James4, John R. Cooley5, Daniel G. Panaccione1, Jørgen Eilenberg6, Henrik H. De Fine Licht6, 6 Angie M. Macias1, Matthew C. Berger1, Kristen L. Wickert1, Cameron M. Stauder1, Ellie J. 7 Spahr1, Matthew D. Maust1, Amy M. Metheny1, Chris Simon5, Gene Kritsky7, Kathie T. Hodge8, 8 Richard A. Humber8,9, Terry Gullion10, Dylan P. G. Short11, Teiya Kijimoto1, Dan Mozgai12, 9 Nidia Arguedas13, Matt T. Kasson1,* 10 11 1Division of Plant and Soil Sciences, West Virginia University, Morgantown, West Virginia, 12 26506, USA. 13 2Department of Plant Pathology, The Ohio State University, Columbus, Ohio 43210, USA. 14 3Department of Microbiology and Plant Pathology and Institute for Integrative Genome Biology, 15 University of California, Riverside, California 92521, USA. 16 4Department of Ecology and Evolution, University of Michigan, Ann Arbor, Michigan 48109, 17 USA. 18 5Department of Ecology and Evolutionary Biology, University of Connecticut, Storrs, 19 Connecticut 06269, USA. 20 6Department of Plant and Environmental Science, University of Copenhagen, Denmark. 21 7Department of Biology, Mount St. Joseph University, Cincinnati, Ohio 45233, USA. 22 8Plant Pathology & Plant-Microbe Biology, School of Integrative Plant Science, Cornell 23 University, Ithaca, New York 14853, USA. -

Resurrection and Emendation of the Hypoxylaceae, Recognised from a Multigene Phylogeny of the Xylariales
Mycol Progress DOI 10.1007/s11557-017-1311-3 ORIGINAL ARTICLE Resurrection and emendation of the Hypoxylaceae, recognised from a multigene phylogeny of the Xylariales Lucile Wendt1,2 & Esteban Benjamin Sir3 & Eric Kuhnert1,2 & Simone Heitkämper1,2 & Christopher Lambert1,2 & Adriana I. Hladki3 & Andrea I. Romero4,5 & J. Jennifer Luangsa-ard6 & Prasert Srikitikulchai6 & Derek Peršoh7 & Marc Stadler1,2 Received: 21 February 2017 /Revised: 12 April 2017 /Accepted: 19 April 2017 # The Author(s) 2017. This article is an open access publication Abstract A multigene phylogeny was constructed, including polymerase II (RPB2), and beta-tubulin (TUB2). Specimens a significant number of representative species of the main were selected based on more than a decade of intensive mor- lineages in the Xylariaceae and four DNA loci the internal phological and chemotaxonomic work, and cautious taxon transcribed spacer region (ITS), the large subunit (LSU) of sampling was performed to cover the major lineages of the the nuclear rDNA, the second largest subunit of the RNA Xylariaceae; however, with emphasis on hypoxyloid species. The comprehensive phylogenetic analysis revealed a clear-cut This article is part of the “Special Issue on ascomycete systematics in segregation of the Xylariaceae into several major clades, honor of Richard P. Korf who died in August 2016”. which was well in accordance with previously established morphological and chemotaxonomic concepts. One of these The present paper is dedicated to Prof. Jack D. Rogers, on the occasion of his fortcoming 80th birthday. clades contained Annulohypoxylon, Hypoxylon, Daldinia,and other related genera that have stromatal pigments and a Section Editor: Teresa Iturriaga and Marc Stadler nodulisporium-like anamorph. -

Microdochium Nivale in Perennial Grasses: Snow Mould Resistance, Pathogenicity and Genetic Diversity
Philosophiae Doctor (PhD), Thesis 2016:32 (PhD), Doctor Philosophiae ISBN: 978-82-575-1324-5 Norwegian University of Life Sciences ISSN: 1894-6402 Faculty of Veterinary Medicine and Biosciences Department of Plant Sciences Philosophiae Doctor (PhD) Thesis 2016:32 Mohamed Abdelhalim Microdochium nivale in perennial grasses: Snow mould resistance, pathogenicity and genetic diversity Microdochium nivale i flerårig gras: Resistens mot snømugg, patogenitet og genetisk diversitet Postboks 5003 Mohamed Abdelhalim NO-1432 Ås, Norway +47 67 23 00 00 www.nmbu.no Microdochium nivale in perennial grasses: Snow mould resistance, pathogenicity and genetic diversity. Microdochium nivale i flerårig gras: Resistens mot snømugg, patogenitet og genetisk diversitet. Philosophiae Doctor (PhD) Thesis Mohamed Abdelhalim Department of Plant Sciences Faculty of Veterinary Medicine and Biosciences Norwegian University of Life Sciences Ås (2016) Thesis number 2016:32 ISSN 1894-6402 ISBN 978-82-575-1324-5 Supervisors: Professor Anne Marte Tronsmo Department of Plant Sciences, Norwegian University of Life Sciences P.O. Box 5003, 1432 Ås, Norway Professor Odd Arne Rognli Department of Plant Sciences, Norwegian University of Life Sciences P.O. Box 5003, 1432 Ås, Norway Adjunct Professor May Bente Brurberg Department of Plant Sciences, Norwegian University of Life Sciences P.O. Box 5003, 1432 Ås, Norway The Norwegian Institute of Bioeconomy Research (NIBIO) Pb 115, NO-1431 Ås, Norway Researcher Dr. Ingerd Skow Hofgaard The Norwegian Institute of Bioeconomy Research (NIBIO) Pb 115, NO-1431 Ås, Norway Dr. Petter Marum Graminor AS. Bjørke forsøksgård, Hommelstadvegen 60 NO-2344 Ilseng, Norway Associate Professor Åshild Ergon Department of Plant Sciences, Norwegian University of Life Sciences P.O. -

Endophytic Species of Xylaria: Cultural and Isozymic Studies
ZOBODAT - www.zobodat.at Zoologisch-Botanische Datenbank/Zoological-Botanical Database Digitale Literatur/Digital Literature Zeitschrift/Journal: Sydowia Jahr/Year: 1993 Band/Volume: 45 Autor(en)/Author(s): Rodrigues K. F., Leuchtmann A., Petrini Orlando Artikel/Article: Endophytic species of Xylaria: cultural and isozymic studies. 116-138 ©Verlag Ferdinand Berger & Söhne Ges.m.b.H., Horn, Austria, download unter www.biologiezentrum.at Endophytic species of Xylaria: cultural and isozymic studies K. F. Rodrigues , A. Leuchtmann & 0. Petrini 'The New York Botanical Garden, Bronx, New York, 10458-5126, USA 2Geobotanisches Institut ETH, Zollikerstrasse 107, CH-8008 Zurich, Switzerland 'Mikrobiologisches Institut, ETH-Zentrum, CH-8092 Zurich, Switzerland Rodrigues, K.F., A. Leuchtmann & O. Petrini (1993). Endophytic species of Xylaria: cultural and isozymic studies. - Sydowia 45 (1): 116-138. Cultural descriptions of endophytic Xylaria species from an Amazonian palm, Euterpe oleracea, are presented. Eighty-one isolates representing 15 species of Xylaria were examined for isozyme variation by means of horizontal starch gel electrophoresis. Results from the isozyme analysis revealed a high degree of intra- and interspecific diversity among Xylaria species. Keywords: endophytes, isozymes, tropical fungi, Xylariaceae, Euterpe oleracea. The genus Xylaria and other members of the Xylariaceae are commonly isolated endophytes. The identification at the species level of putative endophytic Xylaria grown in culture is still a difficult task because they rarely produce morphologically diagnostic structures, and teleomorphs are seldom formed. Indeed, many of them may differ from free-living forms and some might not produce the teleomorph at all (Brunner & Petrini, 1992). Anamorphs produced in culture are re- latively easy to identify to the genus because of their typical stromata, conidiophores and conidial development. -

Characterizing Fungal Decay of Beech Wood: Potential for Biotechnological Applications
microorganisms Article Characterizing Fungal Decay of Beech Wood: Potential for Biotechnological Applications Ehsan Bari 1,* , Katie Ohno 2, Nural Yilgor 3 , Adya P. Singh 4, Jeffrey J. Morrell 5, Antonio Pizzi 6 , Mohammad Ali Tajick Ghanbary 7 and Javier Ribera 8,* 1 Department of Wood Science and Engineering, Section of Wood Microbiology and Genetic, Technical Faculty of No. 1, Mazandaran Branch, Technical and Vocational University (TVU), Sari 4816831168, Iran 2 USDA Forest Service, Forest Products Laboratory, One Gifford Pinchot Drive, Madison, WI 53726, USA; [email protected] 3 Department of Forest Products Chemistry and Technology Division, Forest Industry Engineering, Forestry Faculty, Istanbul University Cerrahpa¸sa,34473 Istanbul, Turkey; [email protected] 4 Scion, Rotorua 3046, New Zealand; [email protected] 5 National Centre for Timber Durability and Design Life, University of the Sunshine Coast, Brisbane 4102, Australia; [email protected] 6 ENSTIB-LERMAB, University of Lorraine, BP 21042, CEDEX 09, 88051 Epinal, France; [email protected] 7 Department of Mycology and Plant Pathology, College of Agronomic Sciences, Sari Agricultural Sciences and Natural Resources University, Sari 4818166996, Iran; [email protected] 8 Laboratory for Cellulose & Wood Materials, Empa-Swiss Federal Laboratories for Materials Science and Technology, CH-9014 St. Gallen, Switzerland * Correspondence: [email protected] (E.B.); [email protected] (J.R.); Tel.: +98-9354367572 (E.B.); +41-587657607 (J.R.) Abstract: The biotechnological potential of nine decay fungi collected from stored beech logs at a pulp and paper factory yard in Northern Iran was investigated. Beech blocks exposed to the Citation: Bari, E.; Ohno, K.; Yilgor, fungi in a laboratory decay test were used to study changes in cell wall chemistry using both wet N.; Singh, A.P.; Morrell, J.J.; Pizzi, A.; chemistry and spectroscopic methods.