NGS in Hereditary Ataxia: When Rare Becomes Frequent
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Clinical Vignette Novel Bi-Allelic Variants in GJC2 Associated
Clinical Vignette Novel Bi-allelic Variants in GJC2 Associated Pelizaeus- Merzbacher-like Disease 1: Clinical Clues and Differential Diagnosis Veronica Arora, Sapna Sandal, Ishwar Verma Institute of Medical Genetics and Genomics, Sir Ganga Ram Hospital, New Delhi Correspondence to: Dr Ishwar C Verma Email: [email protected] Abstract the environment and did not follow objects. Head titubation was present. There was no facial dys- Hypomyelinating Leukodystrophy-2 (HLD2) or morphism. Anthropometric measurements were Pelizaeus-Merzbacher-like disease 1 (PMLD1) is a as follows: length 82cm (+1.2SD), weight 10.6Kg slowly progressive leukodystrophy characterized (+1.1SD) and head circumference 47.7cm (+1.2SD). by nystagmus, hypotonia, and developmental Central nervous system examination showed bilat- delay. It is a close differential diagnosis for eral pendular nystagmus, axial hypotonia, dystonic Pelizaeus- Merzbacher disease (PMD) and should posturing, and choreo-athetoid movements (Figure be suspected in patients with features of PMD but 1). Deep tendon reflexes were brisk with extensor who are negative on testing for duplication of the plantar responses. The rest of the systemic PLP1 gene. We describe a case of a 16-month-old examination was non-contributory. MRI of the boy with a novel homozygous mutation in the GJC2 brain (axial view) showed diffuse hypo-myelination gene resulting in hypomyelinating leukodystrophy- in the peri-ventricular and sub-cortical area and 2. The clinical clues as well as features of other cerebellar white matter changes (Figure 2). disorders presenting similarly are discussed. Given the presence of hypotonia, brisk reflexes, nystagmus and hypomyelination on MRI, a deletion Clinical description duplication analysis for the PLP1 gene was done which was negative. -

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

Caracterización De Las Bases Genéticas De Ataxias Hereditarias
8. Appendices Appendix I: Genetic tests previously performed for patients included in WES analysis and SCA36 screening. These analyses include the most common forms of ADCA and ARCA caused by trinucleotide expansions and a panel containing genes relevant for different types of ataxia. SCA1, SCA2, SCA3, SCA6, SCA7, SCA12, SCA17 and DRPLA (Dentatorubral- pallidoluysian atrophy): caused by CAG triplet expansions. SCA8: caused by CTG triplet expansions. Friedreich’s ataxia: caused by GAA triplet expansions. SureSelect Human All Exon V6Panel (Agilent Technologies, Santa Clara, CA, USA): this panel was used to study both the coding and the intronic flanking regions of ataxia related genes through NGS. Different sets of genes were used for different types of ataxia. - Episodic ataxia: ATP1A2, ATP1A3, CACNA1A, CACNA1S, CACNB4, KCNA1, SCN1A, SCN2A, SCN4A, SLC1A3. - Dominant ataxia: AFG3L2, ATP1A3, CACNA1A, CACNA1G, CACNB4, CCDC88C, DNMT1, EEF2, ELOVL4, ELOVL5, FAT2, FGF12, FGF14, ITPR1,KCNA1, KCNC3, KCND3, PDYN, PRKCG, SCN2A, SLC1A3, SPG7, SPTBN2, TBP, TGM6, TMEM240, TTBK2, TUBB4A, TRPC2, ME, PLD3. - Recessive ataxia: ABHD12, ADCK3, ANO10, APTX, ATCAY, ATM, ATP8A2, C10orf2, CA8, CWF19L1, ADCK3, CYP27A1, DNAJC19, FXN, GRM1,KCNJ10, KIAA0226, MRE11, MTPAP, PIK3R5, PLEKHG4, PMPCA, PNKP, POLG, RNF216, SACS, SETX, SIL1, SNX14, SYNE1, SYT14, TDP1, TPP1, TTPA, VLDLR, WDR81, WWOX - Complex disorders with prominent ataxia (AR): AFG3L2, CLCN2, COX20, CP, DARS2, FLVCR1, HEXA, HEXB, ITPR1, LAMA1, MTTP, NCP1, NCP2, PLA2G6, PM2, PNPLA6, SPTBN2. - Complex disorders with occasional ataxia (AR): ACO2, AHI1, ARL13B, CC2D2A, CLN5, CLN6, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, GOSR2, L2HGDH, OPA1, PEX7, PHYH, POLR3A, POLR3B, PTF1A, SLC17A5, SLC25A46, SLC52A2, SLC6A19, TSFM, TXN2, WFS1. - Disorders reported with ataxia but not included in the differential diagnosis: EPM2A, NHLRC1, MLC1, COL18A1, HSD17B4, PEX2, WFS1, ASPA, ARSA, SLC2A1. -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -
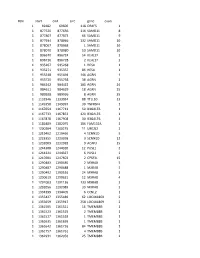
Chr Start End Size Gene Exon 1 69482 69600 118 OR4F5 1 1 877520
#chr start end size gene exon 1 69482 69600 118 OR4F5 1 1 877520 877636 116 SAMD11 8 1 877807 877873 66 SAMD11 9 1 877934 878066 132 SAMD11 10 1 878067 878068 1 SAMD11 10 1 878070 878080 10 SAMD11 10 1 896670 896724 54 KLHL17 2 1 896726 896728 2 KLHL17 2 1 935267 935268 1 HES4 1 1 935271 935357 86 HES4 1 1 955548 955694 146 AGRN 1 1 955720 955758 38 AGRN 1 1 984242 984422 180 AGRN 24 1 984611 984629 18 AGRN 25 1 989928 989936 8 AGRN 35 1 1132946 1133034 88 TTLL10 13 1 1149358 1149397 39 TNFRSF4 1 1 1167654 1167713 59 B3GALT6 1 1 1167733 1167853 120 B3GALT6 1 1 1167878 1167908 30 B3GALT6 1 1 1181889 1182075 186 FAM132A 1 1 1200204 1200215 11 UBE2J2 2 1 1219462 1219466 4 SCNN1D 5 1 1223355 1223358 3 SCNN1D 12 1 1232009 1232018 9 ACAP3 15 1 1244308 1244320 12 PUSL1 2 1 1244321 1244327 6 PUSL1 2 1 1247601 1247603 2 CPSF3L 15 1 1290483 1290485 2 MXRA8 5 1 1290487 1290488 1 MXRA8 5 1 1290492 1290516 24 MXRA8 5 1 1290619 1290631 12 MXRA8 4 1 1291003 1291136 133 MXRA8 3 1 1292056 1292089 33 MXRA8 2 1 1334399 1334405 6 CCNL2 1 1 1355427 1355489 62 LOC441869 2 1 1355659 1355917 258 LOC441869 2 1 1361505 1361521 16 TMEM88B 1 1 1361523 1361525 2 TMEM88B 1 1 1361527 1361528 1 TMEM88B 1 1 1361635 1361636 1 TMEM88B 1 1 1361642 1361726 84 TMEM88B 1 1 1361757 1361761 4 TMEM88B 1 1 1362931 1362956 25 TMEM88B 2 1 1374780 1374838 58 VWA1 3 1 1374841 1374842 1 VWA1 3 1 1374934 1375100 166 VWA1 3 1 1389738 1389747 9 ATAD3C 4 1 1389751 1389816 65 ATAD3C 4 1 1389830 1389849 19 ATAD3C 4 1 1390835 1390837 2 ATAD3C 5 1 1407260 1407331 71 ATAD3B 1 1 1407340 1407474 -

A Translaminar Genetic Logic for the Circuit Identity of Intracortically-Projecting Neurons
bioRxiv preprint doi: https://doi.org/10.1101/290395; this version posted March 28, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title A translaminar genetic logic for the circuit identity of intracortically-projecting neurons Authors Esther Klingler1, Andres De la Rossa1,3, Sabine Fièvre1, Denis Jabaudon1,2 1Department of Basic Neurosciences, University of Geneva, Geneva, Switzerland. 2Clinic of Neurology, Geneva University Hospital, Geneva, Switzerland. 3Present address: Department of Cell Biology, University of Geneva, Geneva, Switzerland. Correspondence should be addressed to D.J. ([email protected]). Abstract Distinct subtypes of intracortically-projecting neurons (ICPN) are present in all layers, allowing propagation of information within and across cortical columns. How the molecular identities of ICPN relate to their defining anatomical and functional properties is unknown. Here we show that the transcriptional identities of ICPN primarily reflect their input-output connectivities rather than their birth dates or laminar positions. Thus, conserved circuit-related transcriptional programs are at play across cortical layers, which may preserve canonical circuit features across development and evolution. bioRxiv preprint doi: https://doi.org/10.1101/290395; this version posted March 28, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Introduction Neurons of the neocortex are organized into six radial layers, which have appeared at different times during evolution, with the superficial layers representing a more recent acquisition. Input to the neocortex predominantly reaches superficial layers (SL, i.e. -

Insurance and Advance Pay Test Requisition
Insurance and Advance Pay Test Requisition (2021) For Specimen Collection Service, Please Fax this Test Requisition to 1.610.271.6085 Client Services is available Monday through Friday from 8:30 AM to 9:00 PM EST at 1.800.394.4493, option 2 Patient Information Patient Name Patient ID# (if available) Date of Birth Sex designated at birth: 9 Male 9 Female Street address City, State, Zip Mobile phone #1 Other Phone #2 Patient email Language spoken if other than English Test and Specimen Information Consult test list for test code and name Test Code: Test Name: Test Code: Test Name: 9 Check if more than 2 tests are ordered. Additional tests should be checked off within the test list ICD-10 Codes (required for billing insurance): Clinical diagnosis: Age at Initial Presentation: Ancestral Background (check all that apply): 9 African 9 Asian: East 9 Asian: Southeast 9 Central/South American 9 Hispanic 9 Native American 9 Ashkenazi Jewish 9 Asian: Indian 9 Caribbean 9 European 9 Middle Eastern 9 Pacific Islander Other: Indications for genetic testing (please check one): 9 Diagnostic (symptomatic) 9 Predictive (asymptomatic) 9 Prenatal* 9 Carrier 9 Family testing/single site Relationship to Proband: If performed at Athena, provide relative’s accession # . If performed at another lab, a copy of the relative’s report is required. Please attach detailed medical records and family history information Specimen Type: Date sample obtained: __________ /__________ /__________ 9 Whole Blood 9 Serum 9 CSF 9 Muscle 9 CVS: Cultured 9 Amniotic Fluid: Cultured 9 Saliva (Not available for all tests) 9 DNA** - tissue source: Concentration ug/ml Was DNA extracted at a CLIA-certified laboratory or a laboratory meeting equivalent requirements (as determined by CAP and/or CMS)? 9 Yes 9 No 9 Other*: If not collected same day as shipped, how was sample stored? 9 Room temp 9 Refrigerated 9 Frozen (-20) 9 Frozen (-80) History of blood transfusion? 9 Yes 9 No Most recent transfusion: __________ /__________ /__________ *Please contact us at 1.800.394.4493, option 2 prior to sending specimens. -

Ion Channels
UC Davis UC Davis Previously Published Works Title THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Permalink https://escholarship.org/uc/item/1442g5hg Journal British journal of pharmacology, 176 Suppl 1(S1) ISSN 0007-1188 Authors Alexander, Stephen PH Mathie, Alistair Peters, John A et al. Publication Date 2019-12-01 DOI 10.1111/bph.14749 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology (2019) 176, S142–S228 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels Stephen PH Alexander1 , Alistair Mathie2 ,JohnAPeters3 , Emma L Veale2 , Jörg Striessnig4 , Eamonn Kelly5, Jane F Armstrong6 , Elena Faccenda6 ,SimonDHarding6 ,AdamJPawson6 , Joanna L Sharman6 , Christopher Southan6 , Jamie A Davies6 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 3Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 4Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, A-6020 Innsbruck, Austria 5School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 6Centre for Discovery Brain Science, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. -

Comparative Transcriptome Profiling of the Human and Mouse Dorsal Root Ganglia: an RNA-Seq-Based Resource for Pain and Sensory Neuroscience Research
bioRxiv preprint doi: https://doi.org/10.1101/165431; this version posted October 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq-based resource for pain and sensory neuroscience research Short Title: Human and mouse DRG comparative transcriptomics Pradipta Ray 1, 2 #, Andrew Torck 1 , Lilyana Quigley 1, Andi Wangzhou 1, Matthew Neiman 1, Chandranshu Rao 1, Tiffany Lam 1, Ji-Young Kim 1, Tae Hoon Kim 2, Michael Q. Zhang 2, Gregory Dussor 1 and Theodore J. Price 1, # 1 The University of Texas at Dallas, School of Behavioral and Brain Sciences 2 The University of Texas at Dallas, Department of Biological Sciences # Corresponding authors Theodore J Price Pradipta Ray School of Behavioral and Brain Sciences School of Behavioral and Brain Sciences The University of Texas at Dallas The University of Texas at Dallas BSB 14.102G BSB 10.608 800 W Campbell Rd 800 W Campbell Rd Richardson TX 75080 Richardson TX 75080 972-883-4311 972-883-7262 [email protected] [email protected] Number of pages: 27 Number of figures: 9 Number of tables: 8 Supplementary Figures: 4 Supplementary Files: 6 Word count: Abstract = 219; Introduction = 457; Discussion = 1094 Conflict of interest: The authors declare no conflicts of interest Patient anonymity and informed consent: Informed consent for human tissue sources were obtained by Anabios, Inc. (San Diego, CA). Human studies: This work was approved by The University of Texas at Dallas Institutional Review Board (MR 15-237). -

Discovery of a Molecular Glue That Enhances Uprmt to Restore
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.17.431525; this version posted February 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Discovery of a molecular glue that enhances UPRmt to restore proteostasis via TRKA-GRB2-EVI1-CRLS1 axis Authors: Li-Feng-Rong Qi1, 2 †, Cheng Qian1, †, Shuai Liu1, 2†, Chao Peng3, 4, Mu Zhang1, Peng Yang1, Ping Wu3, 4, Ping Li1 and Xiaojun Xu1, 2 * † These authors share joint first authorship Running title: Ginsenoside Rg3 reverses Parkinson’s disease model by enhancing mitochondrial UPR Affiliations: 1 State Key Laboratory of Natural Medicines, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. 2 Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. 3. National Facility for Protein Science in Shanghai, Zhangjiang Lab, Shanghai Advanced Research Institute, Chinese Academy of Science, Shanghai 201210, China 4. Shanghai Science Research Center, Chinese Academy of Sciences, Shanghai, 201204, China. Corresponding author: Ping Li, State Key Laboratory of Natural Medicines, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. Email: [email protected], Xiaojun Xu, State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. Telephone number: +86-2583271203, E-mail: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2021.02.17.431525; this version posted February 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Download Positional Information of Genomic Sequence (Refseq Gene: Hg38) and Common Snps (Snp147common: Hg38) in the Region of Interest
Abstracts medgen 2017 · 29:86–192 DOI 10.1007/s11825-017-0126-6 Topics Online publiziert: 23. Februar 2017 © Springer-Verlag Berlin Heidelberg 2017 Keynote Lecture Workshops (W1-9) WS1 Monogenic Disease WS2 Cancer Genetics I Plenary Sessions (PLEN) WS3 Enhancers, Modifiers and Rare Variants Plen 1 WS4 Intellectual Disability Plen 2 WS5 Technology WS6 Complex Genetics WS7 Clinical Genetics Symposia (S1-6) WS8 Cancer Genetics II WS9 Basic Mechanisms and Epigenetics S1 Studying Genetic Diseases with iPSCs and Organoids S2 Intracellular Organelles and their Diseases S3 Huntington Disease Poster (P-) S4 Consequences of Human Germline Variations S6 The Genetic and Epigenetic Basis of Obesity P-BasEpi 001–029 P-Basic Mechanisms and Epigenetic S6 Genomics of Immune Mechanisms P-CancG 030–049 P-Cancer Genetics P-ClinG 050–115 P-Clinical Genetics, Genetic Counselling and Prenatal Diagnosis Selected Presentations (SEL1-4) P-Compl 116–126 P-Complex Diseases, Population & Evolutionary Genetics and Genetic Epidemiology P-CytoG 127–135 P-Cytogenetics and CNVs Sessions P-MonoG 136–169 P-Monogenic Diseases – Educational Session 1 from Gene Identification to Molecular Educational Session 2 Mechanisms Pro & Contra I P-Techno 170–180 P-Technology and Bioinformatics Pro & Contra II P-Therap 181–184 P-Therapy for Genetic Diseases Talk nach 12 DFG-Workshop Oral History Projekt Qualitätsmanagement-Workshop SHG-Session 86 medizinische genetik 1 · 2017 Vorträge Plenary Sessions Keynote Lecture Plen 1 X chromosome structure and regulation Human Olfactory Receptors: Their role in odor perception and as C. M. Disteche new targets for diagnosis and therapy Departments of Pathology and Medicine (Medical Genetics) University of Washington Seattle WA98195, USA H.