Nesprin 1 Is Critical for Nuclear Positioning and Anchorage
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Discovery of a Molecular Glue That Enhances Uprmt to Restore
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.17.431525; this version posted February 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Discovery of a molecular glue that enhances UPRmt to restore proteostasis via TRKA-GRB2-EVI1-CRLS1 axis Authors: Li-Feng-Rong Qi1, 2 †, Cheng Qian1, †, Shuai Liu1, 2†, Chao Peng3, 4, Mu Zhang1, Peng Yang1, Ping Wu3, 4, Ping Li1 and Xiaojun Xu1, 2 * † These authors share joint first authorship Running title: Ginsenoside Rg3 reverses Parkinson’s disease model by enhancing mitochondrial UPR Affiliations: 1 State Key Laboratory of Natural Medicines, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. 2 Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. 3. National Facility for Protein Science in Shanghai, Zhangjiang Lab, Shanghai Advanced Research Institute, Chinese Academy of Science, Shanghai 201210, China 4. Shanghai Science Research Center, Chinese Academy of Sciences, Shanghai, 201204, China. Corresponding author: Ping Li, State Key Laboratory of Natural Medicines, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. Email: [email protected], Xiaojun Xu, State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. Telephone number: +86-2583271203, E-mail: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2021.02.17.431525; this version posted February 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Human Leucine-Rich Repeat Proteins: a Genome-Wide Bioinformatic Categorization and Functional Analysis in Innate Immunity
Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity Aylwin C. Y. Nga,b,1, Jason M. Eisenberga,b,1, Robert J. W. Heatha, Alan Huetta, Cory M. Robinsonc, Gerard J. Nauc, and Ramnik J. Xaviera,b,2 aCenter for Computational and Integrative Biology, and Gastrointestinal Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114; bThe Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02142; and cMicrobiology and Molecular Genetics, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261 Edited by Jeffrey I. Gordon, Washington University School of Medicine, St. Louis, MO, and approved June 11, 2010 (received for review February 17, 2010) In innate immune sensing, the detection of pathogen-associated proteins have been implicated in human diseases to date, notably molecular patterns by recognition receptors typically involve polymorphisms in NOD2 in Crohn disease (8, 9), CIITA in leucine-rich repeats (LRRs). We provide a categorization of 375 rheumatoid arthritis and multiple sclerosis (10), and TLR5 in human LRR-containing proteins, almost half of which lack other Legionnaire disease (11). identifiable functional domains. We clustered human LRR proteins Most LRR domains consist of a chain of between 2 and 45 by first assigning LRRs to LRR classes and then grouping the proteins LRRs (12). Each repeat in turn is typically 20 to 30 residues long based on these class assignments, revealing several of the resulting and can be divided into a highly conserved segment (HCS) fol- protein groups containing a large number of proteins with certain lowed by a variable segment (VS). -

Nuclear Envelope and Muscular Dystrophy
Nationwide Children’s Hospital August 25, 2015 Nuclear Envelope and Muscular Dystrophy Howard J. Worman, M.D. Columbia University Columbia University Medical Center The Nuclear Envelope By D. W. Fawcett The Nuclear Envelope: Nuclear Lamina Dauer & Worman, 2009 Nuclear Lamins: A Brief “Ancient” History Dwyer& Blobel (1975) Aebi et al. (1986) Aebi et al. (1986) Gerace, Blum & Blobel (1978) Goldman et al. (1986) McKeon et al. (1986) Fisher et al. (1986) HUMAN NUCLEAR LAMINS LOCUS CHROMOSOME PROTEINS CELL-TYPES EXPRESSED LMNA 1q21.2-21.3 Lamin A Differentiated Somatic Lamin C Differentiated Somatic Lamin A D10 Differentiated Somatic Lamin C2 Germ LMNB1 5q23.2-31.1 Lamin B1 Apparently All Somatic LMNB2 19p13.3 Lamin B2 All or Most Somatic Lamin B3 Germ LMNA Encoding A-type Lamins Lin and Worman, JBC, 1993 Localization to chromosome 1q21.2–q21.3 Wynder et al., Genomics, 1995 Mutations in LMNA Cause Diseases (“Laminopathies”) with Four Major Tissue-Selective Phenotypes Dauer and Worman Dev. Cell (2009) LMNA Genotype- Phenotype Correlations Dominant mutations causing muscle diseases are missense, splicing and small deletions and even haploinsufficiency Recessive missense mutation peripheral neuropathy Dominant mutations cluster in a small region encoded of exon 8 in partial lipodystrophy Recessive missense in MAD Dominant G608G or S608G in HGPF Worman and Courvalin (2005) Hutchinson-Gilford Progeria Syndrome (HGPS) Normally, lamin A starts as a precursor prelamin A, which is fanesylated. The farnesylated precursor is recognized by ZMPSTE24 endoprotease and cleaved; mature lamin A is not farnesylated. From Worman and Courvalin. (2005) farnesyl The LMNA mutation causing HGPS leads to expression of Paradisi et al. -

The Genomics of Ecological Flexibility, Large Brains, and Long Lives in Capuchin Monkeys Revealed with Fe- Calfacs,” by Joseph D
Correction ANTHROPOLOGY Correction for “The genomics of ecological flexibility, large brains, and long lives in capuchin monkeys revealed with fe- calFACS,” by Joseph D. Orkin, Michael J. Montague, Daniela Tejada-Martinez, Marc de Manuel, Javier del Campo, Saul Cheves Hernandez, Anthony Di Fiore, Claudia Fontsere, Jason A. Hodgson, Mareike C. Janiak, Lukas F. K. Kuderna, Esther Lizano, Maria Pia Martin, Yoshihito Niimura, George H. Perry, Carmen Soto Valverde, Jia Tang, Wesley C. Warren, João Pedro de Magalhães, Shoji Kawamura, Tomàs Marquès-Bonet, Roman Krawetz, and Amanda D. Melin, which published February 11, 2021; 10.1073/ pnas.2010632118 (Proc. Natl. Acad. Sci. U.S.A. 118, e2010632118). The authors note that Fig. 4 in this article uses previously published silhouette images for depicted species. This Correction updates the caption to reflect the necessary credit lines for the material reused. The updated caption is: “Genes under positive selection in white-faced capuchin monkeys show enrichment associated with longevity (maximum recorded lifespan in cap- tivity) and brain size and development. Several genes showing evidence of positive selection in the Cebus lineage are listed for CORRECTION each trait. The displayed species are the primates used for the PAML run, or a congeneric species in cases of missing trait data (e.g., C. capucinus in place of C. imitator). Relative brain size, calculated as EQ = brain mass/(0.085 × (body mass0.775)) (1), is displayed to account for the large range in body mass. Trait data are from refs. 1, 8, 109, and 110. Cebus capucinus silhouette credit: Phylopic/Sarah Werning, licensed under CC BY 3.0. -

Targeted Next-Generation Sequencing Identified a Known EMD Mutation In
Dai et al. Human Genome Variation (2019) 6:42 https://doi.org/10.1038/s41439-019-0072-8 Human Genome Variation DATA REPORT Open Access Targeted next-generation sequencing identified a known EMD mutation in a Chinese patient with Emery-Dreifuss muscular dystrophy Xiafei Dai1,2, Chenqing Zheng3,XuepinChen2,4, Yibin Tang2, Hongmei Zhang2,ChaoYan1,2,HuihuiMa1,2 and Xiaoping Li1,2 Abstract Emery-Dreifuss muscular dystrophy (EDMD) is a rare X-linked recessive disease characterized by the clinical triad of early childhood joint contractures, progressive weakness in muscles and cardiac involvement and can result in sudden death. Targeted next-generation sequencing was performed for a Chinese patient with EDMD and the previously reported mutation [NM_000117.2: c.251_255del (p.Leu84Profs*7)] in exon 3 of the emerin gene (EMD) was identified. Emery-Dreifuss muscular dystrophy (EDMD) is a rare EDMD)13 and autosomal dominant EDMD (AD- progressive neuromuscular disease. It is characterized by EDMD)14 were also reported15. Usually, X-EDMD is 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; the clinical trial of (1) early-onset contractures of the milder and rarer than AD-EDMD. elbows, Achilles tendons and posterior neck; (2) slowly A pedigree comprising 5 members from Hubei Pro- progressive muscle wasting and weakness; and (3) cardi- vince, China, was enrolled in our study. This study was omyopathy with cardiac conduction defects, resulting in approved by the Sichuan Academy of Medical Sciences – high-risk sudden death1 5. Genetically, EDMD has been and Sichuan Provincial People’s Hospital Trust Ethics associated with EMD, LMNA, FHL1, SYNE1, SYNE2, Committee. Written informed consent was obtained from LUMA, and SUN1 gene mutations; the most common all participants. -
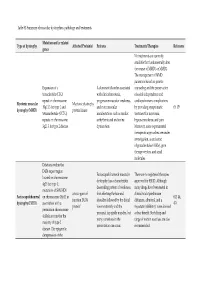
PDF-Document
Table S1.Summary of muscular dystrophies, pathology and treatments. Mutation and/or related Type of dystrophy Affected Protein(s) Features Treatments/Therapies Reference genes No treatments are currently available that fundamentally alter the course of MMD1 or MMD2. The management of MMD patients is based on genetic Expansion of a A dominant disorder associated counseling and the preservation trinucleotide (CTG) with clinical myotonia, of social independence and repeat on chromosome progressive muscular weakness, cardiopulmonary complications Myotonic muscular Myotonic dystrophy 19q13.3 for type 1 and and extra muscular by providing symptomatic (9, 15) dystrophy (MMD) protein kinase tetranucleotide (CCTG) manifestations such as cardiac treatment for myotonia, repeats on chromosome arrhythmia and endocrine hypersomnolence, and pain. 3q21.3 for type 2 disease. dysfunction. Moreover, some experimental therapeutic approaches are under investigation, as antisense oligonucleotides (ASOs), gene therapy vectors, and small molecules. Deletions within the D4Z4 repeat region Facioscapulohumeral muscular There are no registered therapies located on chromosome dystrophy has a characteristic approved for FSHD. Although 4q35 for type 1; descending pattern of weakness, many drugs have been tested in mutations of SMCHD1 a toxic-gain-of- first affecting the face and clinical trials (prednisone Facioscapulohumeral on chromosome 18p11 in (43, 44, function DUX4 shoulder followed by the distal diltiazem, albuterol, and a dystrophy (FSHD) association with a 47) protein’ lower extremity and the myostatin inhibitor), none showed permission chromosome proximal hip girdle muscles, but a clear benefit. Stretching and 4 allele account for the many variations in the range of motion exercises are also majority of type 2 presentation can occur. -

Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss
cells Review Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss 1, 1, 1 1,2,3 1 Gayle B. Collin y, Navdeep Gogna y, Bo Chang , Nattaya Damkham , Jai Pinkney , Lillian F. Hyde 1, Lisa Stone 1 , Jürgen K. Naggert 1 , Patsy M. Nishina 1,* and Mark P. Krebs 1,* 1 The Jackson Laboratory, Bar Harbor, Maine, ME 04609, USA; [email protected] (G.B.C.); [email protected] (N.G.); [email protected] (B.C.); [email protected] (N.D.); [email protected] (J.P.); [email protected] (L.F.H.); [email protected] (L.S.); [email protected] (J.K.N.) 2 Department of Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand 3 Siriraj Center of Excellence for Stem Cell Research, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand * Correspondence: [email protected] (P.M.N.); [email protected] (M.P.K.); Tel.: +1-207-2886-383 (P.M.N.); +1-207-2886-000 (M.P.K.) These authors contributed equally to this work. y Received: 29 February 2020; Accepted: 7 April 2020; Published: 10 April 2020 Abstract: Inherited retinal degeneration (RD) leads to the impairment or loss of vision in millions of individuals worldwide, most frequently due to the loss of photoreceptor (PR) cells. Animal models, particularly the laboratory mouse, have been used to understand the pathogenic mechanisms that underlie PR cell loss and to explore therapies that may prevent, delay, or reverse RD. Here, we reviewed entries in the Mouse Genome Informatics and PubMed databases to compile a comprehensive list of monogenic mouse models in which PR cell loss is demonstrated. -

Biallelic SYNE2 Missense Mutations Leading to Nesprin-2 Giant Hypo-Expression Are Associated with Intellectual Disability and Autism
G C A T T A C G G C A T genes Article Biallelic SYNE2 Missense Mutations Leading to Nesprin-2 Giant Hypo-Expression Are Associated with Intellectual Disability and Autism Natalie Young 1,†, Maria Asif 2,3,†, Matthew Jackson 1 , Daniel Martín Fernández-Mayoralas 4, Mar Jimenez de la Peña 4, Beatriz Calleja-Pérez 5, Sara Álvarez 6, Eve Hunter-Featherstone 1, Angelika A. Noegel 3 , Wolfgang Höhne 2, Peter Nürnberg 2,7 , Boguslaw Obara 8,9, Muhammad Sajid Hussain 2,3,7,* , Iakowos Karakesisoglou 1,* and Alberto Fernández-Jaén 10,* 1 Department of Biosciences, University of Durham, South Road, Durham DH1 3LE, UK; [email protected] (N.Y.); [email protected] (M.J.); [email protected] (E.H.-F.) 2 Cologne Center for Genomics (CCG), University Hospital Cologne, University of Cologne, 50931 Cologne, Germany; [email protected] (M.A.); [email protected] (W.H.); [email protected] (P.N.) 3 Center for Biochemistry, Medical Faculty, University of Cologne, 50931 Cologne, Germany; [email protected] 4 Department of Pediatric Neurology, Hospital Universitario Quirónsalud, 28223 Madrid, Spain; [email protected] (D.M.F.-M.); [email protected] (M.J.d.l.P.) 5 Citation: Young, N.; Asif, M.; Pediatric Primary Care, C. S. Doctor Cirajas, 28017 Madrid, Spain; [email protected] 6 Department of Genomics and Medicine, Genomics and Medicine, NIMGenetics, 28108 Madrid, Spain; Jackson, M.; Fernández-Mayoralas, [email protected] D.M.; de la Peña, M.J.; Calleja-Pérez, 7 Center for Molecular Medicine Cologne (CMMC), University Hospital Cologne, University of Cologne, B.; Álvarez, S.; Hunter-Featherstone, 50931 Cologne, Germany E.; Noegel, A.A.; Höhne, W.; et al. -

Maintains Nuclear Envelope Architecture and Composition in Skin
Research Article 1887 Nesprin-2 Giant (NUANCE) maintains nuclear envelope architecture and composition in skin Yvonne Lüke1,*, Hafida Zaim1,*, Iakowos Karakesisoglou1,2,*, Verena M. Jaeger1, Lorenz Sellin3, Wenshu Lu1, Maria Schneider1, Sascha Neumann1,4, Asa Beijer1, Martina Munck1,4, V. C. Padmakumar1,4, Joachim Gloy3, Gerd Walz3 and Angelika A. Noegel1,4,‡ 1Center for Biochemistry, Medical Faculty, University of Cologne, Joseph-Stelzmann-Str. 52, 50931 Cologne, Germany 2Department of Biological Sciences, The School of Biological and Biomedical Sciences, The University of Durham, Durham DH1 3LE, UK 3Renal Division, University Hospital Freiburg, 79106 Freiburg, Germany 4Center for Molecular Medicine Cologne (CMMC) and Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne, Joseph-Stelzmann-Strasse 52, 50931 Cologne, Germany *These authors contributed equally to this work ‡Author for correspondence (e-mail: [email protected]) Accepted 20 March 2008 Journal of Cell Science 121, 1887-1898 Published by The Company of Biologists 2008 doi:10.1242/jcs.019075 Summary Giant isoforms, encoded by Nesprin-1 (Syne1) and Nesprin-2 the nuclear envelope in mutant fibroblasts, often forming (Syne2), are multifunctional actin-binding and nuclear- aggregates in the deformed nuclear envelope areas. Thus, envelope-associated proteins belonging to the spectrin Nesprin-2 is an important scaffold protein implicated in the superfamily. Here, we investigate the function of Nesprin-2 maintenance of nuclear envelope architecture. Aged knockout Giant (NUANCE) in skin by generating mice lacking the actin- fibroblasts readily generated, by alternative splicing and binding domain of Nesprin-2 (Nesprin-2ΔABD). This loss results alternative translation initiation, aberrant Nesprin-2 Giant in a slight but significant thickening of the epidermis, which is isoforms that lacked an ABD but that were sufficient to restore a consequence of the increased epithelial nuclear size. -

NGS in Hereditary Ataxia: When Rare Becomes Frequent
International Journal of Molecular Sciences Article NGS in Hereditary Ataxia: When Rare Becomes Frequent Daniele Galatolo 1, Giovanna De Michele 2, Gabriella Silvestri 3,4 , Vincenzo Leuzzi 5 , Carlo Casali 6, Olimpia Musumeci 7 , Antonella Antenora 2, Guja Astrea 1, Melissa Barghigiani 1, Roberta Battini 1 , Carla Battisti 8, Caterina Caputi 5, Ettore Cioffi 6, Giuseppe De Michele 2, Maria Teresa Dotti 8, Tommasina Fico 2, Chiara Fiorillo 9, Serena Galosi 5, Maria Lieto 2, Alessandro Malandrini 8, Marina A. B. Melone 10 , Andrea Mignarri 8, Gemma Natale 1, Elena Pegoraro 11, Antonio Petrucci 12 , Ivana Ricca 1 , Vittorio Riso 3,4 , Salvatore Rossi 3,4, Anna Rubegni 1 , Arianna Scarlatti 1, Francesca Tinelli 1, Rosanna Trovato 1, Gioacchino Tedeschi 10, Alessandra Tessa 1,*, Alessandro Filla 2,* and Filippo Maria Santorelli 1,* 1 Molecular Medicine for Neurodegenerative and Neuromuscular Diseases Unit, IRCCS Stella Maris Foundation, 56128 Pisa, Italy; [email protected] (D.G.); [email protected] (G.A.); [email protected] (M.B.); [email protected] (R.B.); [email protected] (G.N.); [email protected] (I.R.); [email protected] (A.R.); [email protected] (A.S.); [email protected] (F.T.); [email protected] (R.T.) 2 Department of Neurosciences, Reproductive and Odontostomatological Sciences, Federico II University, 80131 Naples, Italy; [email protected] (G.D.M.); [email protected] (A.A.); [email protected] (G.D.M.); fi[email protected] (T.F.); [email protected]