A Translaminar Genetic Logic for the Circuit Identity of Intracortically-Projecting Neurons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Autism Multiplex Family with 16P11.2P12.2 Microduplication Syndrome in Monozygotic Twins and Distal 16P11.2 Deletion in Their Brother
European Journal of Human Genetics (2012) 20, 540–546 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Autism multiplex family with 16p11.2p12.2 microduplication syndrome in monozygotic twins and distal 16p11.2 deletion in their brother Anne-Claude Tabet1,2,3,4, Marion Pilorge2,3,4, Richard Delorme5,6,Fre´de´rique Amsellem5,6, Jean-Marc Pinard7, Marion Leboyer6,8,9, Alain Verloes10, Brigitte Benzacken1,11,12 and Catalina Betancur*,2,3,4 The pericentromeric region of chromosome 16p is rich in segmental duplications that predispose to rearrangements through non-allelic homologous recombination. Several recurrent copy number variations have been described recently in chromosome 16p. 16p11.2 rearrangements (29.5–30.1 Mb) are associated with autism, intellectual disability (ID) and other neurodevelopmental disorders. Another recognizable but less common microdeletion syndrome in 16p11.2p12.2 (21.4 to 28.5–30.1 Mb) has been described in six individuals with ID, whereas apparently reciprocal duplications, studied by standard cytogenetic and fluorescence in situ hybridization techniques, have been reported in three patients with autism spectrum disorders. Here, we report a multiplex family with three boys affected with autism, including two monozygotic twins carrying a de novo 16p11.2p12.2 duplication of 8.95 Mb (21.28–30.23 Mb) characterized by single-nucleotide polymorphism array, encompassing both the 16p11.2 and 16p11.2p12.2 regions. The twins exhibited autism, severe ID, and dysmorphic features, including a triangular face, deep-set eyes, large and prominent nasal bridge, and tall, slender build. The eldest brother presented with autism, mild ID, early-onset obesity and normal craniofacial features, and carried a smaller, overlapping 16p11.2 microdeletion of 847 kb (28.40–29.25 Mb), inherited from his apparently healthy father. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Clinical Vignette Novel Bi-Allelic Variants in GJC2 Associated
Clinical Vignette Novel Bi-allelic Variants in GJC2 Associated Pelizaeus- Merzbacher-like Disease 1: Clinical Clues and Differential Diagnosis Veronica Arora, Sapna Sandal, Ishwar Verma Institute of Medical Genetics and Genomics, Sir Ganga Ram Hospital, New Delhi Correspondence to: Dr Ishwar C Verma Email: [email protected] Abstract the environment and did not follow objects. Head titubation was present. There was no facial dys- Hypomyelinating Leukodystrophy-2 (HLD2) or morphism. Anthropometric measurements were Pelizaeus-Merzbacher-like disease 1 (PMLD1) is a as follows: length 82cm (+1.2SD), weight 10.6Kg slowly progressive leukodystrophy characterized (+1.1SD) and head circumference 47.7cm (+1.2SD). by nystagmus, hypotonia, and developmental Central nervous system examination showed bilat- delay. It is a close differential diagnosis for eral pendular nystagmus, axial hypotonia, dystonic Pelizaeus- Merzbacher disease (PMD) and should posturing, and choreo-athetoid movements (Figure be suspected in patients with features of PMD but 1). Deep tendon reflexes were brisk with extensor who are negative on testing for duplication of the plantar responses. The rest of the systemic PLP1 gene. We describe a case of a 16-month-old examination was non-contributory. MRI of the boy with a novel homozygous mutation in the GJC2 brain (axial view) showed diffuse hypo-myelination gene resulting in hypomyelinating leukodystrophy- in the peri-ventricular and sub-cortical area and 2. The clinical clues as well as features of other cerebellar white matter changes (Figure 2). disorders presenting similarly are discussed. Given the presence of hypotonia, brisk reflexes, nystagmus and hypomyelination on MRI, a deletion Clinical description duplication analysis for the PLP1 gene was done which was negative. -

Transcriptomic Analysis of Native Versus Cultured Human and Mouse Dorsal Root Ganglia Focused on Pharmacological Targets Short
bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Transcriptomic analysis of native versus cultured human and mouse dorsal root ganglia focused on pharmacological targets Short title: Comparative transcriptomics of acutely dissected versus cultured DRGs Andi Wangzhou1, Lisa A. McIlvried2, Candler Paige1, Paulino Barragan-Iglesias1, Carolyn A. Guzman1, Gregory Dussor1, Pradipta R. Ray1,#, Robert W. Gereau IV2, # and Theodore J. Price1, # 1The University of Texas at Dallas, School of Behavioral and Brain Sciences and Center for Advanced Pain Studies, 800 W Campbell Rd. Richardson, TX, 75080, USA 2Washington University Pain Center and Department of Anesthesiology, Washington University School of Medicine # corresponding authors [email protected], [email protected] and [email protected] Funding: NIH grants T32DA007261 (LM); NS065926 and NS102161 (TJP); NS106953 and NS042595 (RWG). The authors declare no conflicts of interest Author Contributions Conceived of the Project: PRR, RWG IV and TJP Performed Experiments: AW, LAM, CP, PB-I Supervised Experiments: GD, RWG IV, TJP Analyzed Data: AW, LAM, CP, CAG, PRR Supervised Bioinformatics Analysis: PRR Drew Figures: AW, PRR Wrote and Edited Manuscript: AW, LAM, CP, GD, PRR, RWG IV, TJP All authors approved the final version of the manuscript. 1 bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Altered Stress-Induced Regulation of Genes in Monocytes in Adults with a History of Childhood Adversity
Neuropsychopharmacology (2016) 41, 2530–2540 © 2016 American College of Neuropsychopharmacology. All rights reserved 0893-133X/16 www.neuropsychopharmacology.org Altered Stress-Induced Regulation of Genes in Monocytes in Adults with a History of Childhood Adversity Marion Schwaiger1, Marianna Grinberg2, Dirk Moser1, Johannes CS Zang1, Markus Heinrichs3,4, Jan G Hengstler5, Jörg Rahnenführer2, Steve Cole6 and Robert Kumsta*,1 1 2 Department of Genetic Psychology, Faculty of Psychology, Ruhr-University Bochum, Bochum, Germany; Department of Statistics, TU Dortmund University, Dortmund, Germany; 3Department of Psychology, Laboratory for Biological and Personality Psychology, University of Freiburg, Freiburg, Germany; 4Freiburg Brain Imaging Center, University Medical Center, University of Freiburg, Freiburg, Germany; 5Leibniz Research Centre for 6 Working Environment and Human Factors at the Technical University of Dortmund (IfADo), Dortmund, Germany; David Geffen School of Medicine, University of California, Los Angeles, CA, USA Exposure to serious or traumatic events early in life can lead to persistent alterations in physiological stress response systems, including enhanced cross talk between the neuroendocrine and immune system. These programming effects may be mechanistically involved in mediating the effects of adverse childhood experience on disease risk in adulthood. We investigated hormonal and genome-wide mRNA = expression responses in monocytes to acute stress exposure, in a sample of healthy adults (n 30) with a history of early childhood = adversity, and a control group (n 30) without trauma experience. The early adversity group showed altered hypothalamus-pituitary- adrenal axis responses to stress, evidenced by lower ACTH and cortisol responses. Analyses of gene expression patterns showed that stress-responsive transcripts were enriched for genes involved in cytokine activity, cytokine–cytokine receptor interaction, chemokine activity, and G-protein coupled receptor binding. -

Caracterización De Las Bases Genéticas De Ataxias Hereditarias
8. Appendices Appendix I: Genetic tests previously performed for patients included in WES analysis and SCA36 screening. These analyses include the most common forms of ADCA and ARCA caused by trinucleotide expansions and a panel containing genes relevant for different types of ataxia. SCA1, SCA2, SCA3, SCA6, SCA7, SCA12, SCA17 and DRPLA (Dentatorubral- pallidoluysian atrophy): caused by CAG triplet expansions. SCA8: caused by CTG triplet expansions. Friedreich’s ataxia: caused by GAA triplet expansions. SureSelect Human All Exon V6Panel (Agilent Technologies, Santa Clara, CA, USA): this panel was used to study both the coding and the intronic flanking regions of ataxia related genes through NGS. Different sets of genes were used for different types of ataxia. - Episodic ataxia: ATP1A2, ATP1A3, CACNA1A, CACNA1S, CACNB4, KCNA1, SCN1A, SCN2A, SCN4A, SLC1A3. - Dominant ataxia: AFG3L2, ATP1A3, CACNA1A, CACNA1G, CACNB4, CCDC88C, DNMT1, EEF2, ELOVL4, ELOVL5, FAT2, FGF12, FGF14, ITPR1,KCNA1, KCNC3, KCND3, PDYN, PRKCG, SCN2A, SLC1A3, SPG7, SPTBN2, TBP, TGM6, TMEM240, TTBK2, TUBB4A, TRPC2, ME, PLD3. - Recessive ataxia: ABHD12, ADCK3, ANO10, APTX, ATCAY, ATM, ATP8A2, C10orf2, CA8, CWF19L1, ADCK3, CYP27A1, DNAJC19, FXN, GRM1,KCNJ10, KIAA0226, MRE11, MTPAP, PIK3R5, PLEKHG4, PMPCA, PNKP, POLG, RNF216, SACS, SETX, SIL1, SNX14, SYNE1, SYT14, TDP1, TPP1, TTPA, VLDLR, WDR81, WWOX - Complex disorders with prominent ataxia (AR): AFG3L2, CLCN2, COX20, CP, DARS2, FLVCR1, HEXA, HEXB, ITPR1, LAMA1, MTTP, NCP1, NCP2, PLA2G6, PM2, PNPLA6, SPTBN2. - Complex disorders with occasional ataxia (AR): ACO2, AHI1, ARL13B, CC2D2A, CLN5, CLN6, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, GOSR2, L2HGDH, OPA1, PEX7, PHYH, POLR3A, POLR3B, PTF1A, SLC17A5, SLC25A46, SLC52A2, SLC6A19, TSFM, TXN2, WFS1. - Disorders reported with ataxia but not included in the differential diagnosis: EPM2A, NHLRC1, MLC1, COL18A1, HSD17B4, PEX2, WFS1, ASPA, ARSA, SLC2A1. -

Absence of S100A4 in the Mouse Lens Induces an Aberrant Retina-Specific Differentiation Program and Cataract
www.nature.com/scientificreports OPEN Absence of S100A4 in the mouse lens induces an aberrant retina‑specifc diferentiation program and cataract Rupalatha Maddala1*, Junyuan Gao2, Richard T. Mathias2, Tylor R. Lewis1, Vadim Y. Arshavsky1,3, Adriana Levine4, Jonathan M. Backer4,5, Anne R. Bresnick4 & Ponugoti V. Rao1,3* S100A4, a member of the S100 family of multifunctional calcium‑binding proteins, participates in several physiological and pathological processes. In this study, we demonstrate that S100A4 expression is robustly induced in diferentiating fber cells of the ocular lens and that S100A4 (−/−) knockout mice develop late‑onset cortical cataracts. Transcriptome profling of lenses from S100A4 (−/−) mice revealed a robust increase in the expression of multiple photoreceptor‑ and Müller glia‑specifc genes, as well as the olfactory sensory neuron‑specifc gene, S100A5. This aberrant transcriptional profle is characterized by corresponding increases in the levels of proteins encoded by the aberrantly upregulated genes. Ingenuity pathway network and curated pathway analyses of diferentially expressed genes in S100A4 (−/−) lenses identifed Crx and Nrl transcription factors as the most signifcant upstream regulators, and revealed that many of the upregulated genes possess promoters containing a high‑density of CpG islands bearing trimethylation marks at histone H3K27 and/or H3K4, respectively. In support of this fnding, we further documented that S100A4 (−/−) knockout lenses have altered levels of trimethylated H3K27 and H3K4. Taken together, -

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. -

PRODUCT SPECIFICATION Anti-GJC3 Product Datasheet
Anti-GJC3 Product Datasheet Polyclonal Antibody PRODUCT SPECIFICATION Product Name Anti-GJC3 Product Number HPA015024 Gene Description gap junction protein, gamma 3, 30.2kDa Clonality Polyclonal Isotype IgG Host Rabbit Antigen Sequence Recombinant Protein Epitope Signature Tag (PrEST) antigen sequence: RTWKHKSSSSKYFLTSESTRRHKKATDSLPVVETKEQFQEAVPGRSLAQE KQRPVGPRDA Purification Method Affinity purified using the PrEST antigen as affinity ligand Verified Species Human Reactivity Recommended IHC (Immunohistochemistry) Applications - Antibody dilution: 1:20 - 1:50 - Retrieval method: HIER pH6 WB (Western Blot) - Working concentration: 0.04-0.4 µg/ml Characterization Data Available at atlasantibodies.com/products/HPA015024 Buffer 40% glycerol and PBS (pH 7.2). 0.02% sodium azide is added as preservative. Concentration Lot dependent Storage Store at +4°C for short term storage. Long time storage is recommended at -20°C. Notes Gently mix before use. Optimal concentrations and conditions for each application should be determined by the user. For protocols, additional product information, such as images and references, see atlasantibodies.com. Product of Sweden. For research use only. Not intended for pharmaceutical development, diagnostic, therapeutic or any in vivo use. No products from Atlas Antibodies may be resold, modified for resale or used to manufacture commercial products without prior written approval from Atlas Antibodies AB. Warranty: The products supplied by Atlas Antibodies are warranted to meet stated product specifications and to conform to label descriptions when used and stored properly. Unless otherwise stated, this warranty is limited to one year from date of sales for products used, handled and stored according to Atlas Antibodies AB's instructions. Atlas Antibodies AB's sole liability is limited to replacement of the product or refund of the purchase price. -
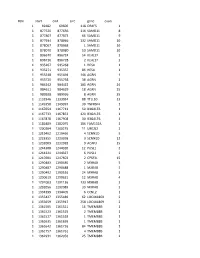
Chr Start End Size Gene Exon 1 69482 69600 118 OR4F5 1 1 877520
#chr start end size gene exon 1 69482 69600 118 OR4F5 1 1 877520 877636 116 SAMD11 8 1 877807 877873 66 SAMD11 9 1 877934 878066 132 SAMD11 10 1 878067 878068 1 SAMD11 10 1 878070 878080 10 SAMD11 10 1 896670 896724 54 KLHL17 2 1 896726 896728 2 KLHL17 2 1 935267 935268 1 HES4 1 1 935271 935357 86 HES4 1 1 955548 955694 146 AGRN 1 1 955720 955758 38 AGRN 1 1 984242 984422 180 AGRN 24 1 984611 984629 18 AGRN 25 1 989928 989936 8 AGRN 35 1 1132946 1133034 88 TTLL10 13 1 1149358 1149397 39 TNFRSF4 1 1 1167654 1167713 59 B3GALT6 1 1 1167733 1167853 120 B3GALT6 1 1 1167878 1167908 30 B3GALT6 1 1 1181889 1182075 186 FAM132A 1 1 1200204 1200215 11 UBE2J2 2 1 1219462 1219466 4 SCNN1D 5 1 1223355 1223358 3 SCNN1D 12 1 1232009 1232018 9 ACAP3 15 1 1244308 1244320 12 PUSL1 2 1 1244321 1244327 6 PUSL1 2 1 1247601 1247603 2 CPSF3L 15 1 1290483 1290485 2 MXRA8 5 1 1290487 1290488 1 MXRA8 5 1 1290492 1290516 24 MXRA8 5 1 1290619 1290631 12 MXRA8 4 1 1291003 1291136 133 MXRA8 3 1 1292056 1292089 33 MXRA8 2 1 1334399 1334405 6 CCNL2 1 1 1355427 1355489 62 LOC441869 2 1 1355659 1355917 258 LOC441869 2 1 1361505 1361521 16 TMEM88B 1 1 1361523 1361525 2 TMEM88B 1 1 1361527 1361528 1 TMEM88B 1 1 1361635 1361636 1 TMEM88B 1 1 1361642 1361726 84 TMEM88B 1 1 1361757 1361761 4 TMEM88B 1 1 1362931 1362956 25 TMEM88B 2 1 1374780 1374838 58 VWA1 3 1 1374841 1374842 1 VWA1 3 1 1374934 1375100 166 VWA1 3 1 1389738 1389747 9 ATAD3C 4 1 1389751 1389816 65 ATAD3C 4 1 1389830 1389849 19 ATAD3C 4 1 1390835 1390837 2 ATAD3C 5 1 1407260 1407331 71 ATAD3B 1 1 1407340 1407474 -

Insurance and Advance Pay Test Requisition
Insurance and Advance Pay Test Requisition (2021) For Specimen Collection Service, Please Fax this Test Requisition to 1.610.271.6085 Client Services is available Monday through Friday from 8:30 AM to 9:00 PM EST at 1.800.394.4493, option 2 Patient Information Patient Name Patient ID# (if available) Date of Birth Sex designated at birth: 9 Male 9 Female Street address City, State, Zip Mobile phone #1 Other Phone #2 Patient email Language spoken if other than English Test and Specimen Information Consult test list for test code and name Test Code: Test Name: Test Code: Test Name: 9 Check if more than 2 tests are ordered. Additional tests should be checked off within the test list ICD-10 Codes (required for billing insurance): Clinical diagnosis: Age at Initial Presentation: Ancestral Background (check all that apply): 9 African 9 Asian: East 9 Asian: Southeast 9 Central/South American 9 Hispanic 9 Native American 9 Ashkenazi Jewish 9 Asian: Indian 9 Caribbean 9 European 9 Middle Eastern 9 Pacific Islander Other: Indications for genetic testing (please check one): 9 Diagnostic (symptomatic) 9 Predictive (asymptomatic) 9 Prenatal* 9 Carrier 9 Family testing/single site Relationship to Proband: If performed at Athena, provide relative’s accession # . If performed at another lab, a copy of the relative’s report is required. Please attach detailed medical records and family history information Specimen Type: Date sample obtained: __________ /__________ /__________ 9 Whole Blood 9 Serum 9 CSF 9 Muscle 9 CVS: Cultured 9 Amniotic Fluid: Cultured 9 Saliva (Not available for all tests) 9 DNA** - tissue source: Concentration ug/ml Was DNA extracted at a CLIA-certified laboratory or a laboratory meeting equivalent requirements (as determined by CAP and/or CMS)? 9 Yes 9 No 9 Other*: If not collected same day as shipped, how was sample stored? 9 Room temp 9 Refrigerated 9 Frozen (-20) 9 Frozen (-80) History of blood transfusion? 9 Yes 9 No Most recent transfusion: __________ /__________ /__________ *Please contact us at 1.800.394.4493, option 2 prior to sending specimens. -

Gene Expression Profiles of Patients with Cerebral Hematoma Following Spontaneous Intracerebral Hemorrhage
MOLECULAR MEDICINE REPORTS 10: 1671-1678, 2014 Gene expression profiles of patients with cerebral hematoma following spontaneous intracerebral hemorrhage TAO YANG1,2, JIANWEN GU1, BIN KONG1,2, YONGQIN KUANG1, LIN CHENG1, JINGMIN CHENG1,2, XUN XIA1,2, YUAN MA1,2 and JUNHAI ZHANG1 1Department of Neurosurgery, Chengdu Military General Hospital, Chengdu, Sichuan 610083; 2Department of Postgraduate, Third Military Medical University, Chongqing 400038, P.R. China Received October 13, 2013; Accepted March 27, 2014 DOI: 10.3892/mmr.2014.2421 Abstract. The present study aimed to investigate the gene ITPR1-RYR2, CAMK2A-RYR2 and ITGA5-LAMB1 interac- functions and expression profiles in perihematomal (PH) tion pairs. The present study provides several potential targets brain regions following spontaneous intracerebral hemor- to decrease hematoma expansion and alleviate neuronal cell rhage. The gene expression profiles were downloaded from death following spontaneous intracerebral hemorrhage. the Gene Expression Omnibus database under accession number GSE24265, which includes 11 brain samples from Introduction different regions, including four samples from PH areas, four from contralateral grey matter (CG) and three from contra- Spontaneous intracerebral hemorrhage (ICH) involves the lateral white matter (CW). The gene expression profiles were extravasation of blood within the brain parenchyma in the pre-processed and the differentially expressed genes (DEGs) absence of trauma or surgery, accounting for 10-30% of all between PH and CG tissue, and PH and CW tissue were identi- strokes worldwide (1). ICH is a complex, dynamic process fied using R packages. The expression of genes in different that consists of three distinct phases (2): i) Initial hemorrhage, tissues was analyzed by hierarchical clustering.