Germline Mutations Affecting the Histone H4 Core Cause a Developmental Syndrome by Altering DNA Damage Response and Cell Cycle Control
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
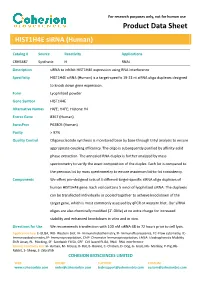
Product Data Sheet
For research purposes only, not for human use Product Data Sheet HIST1H4E siRNA (Human) Catalog # Source Reactivity Applications CRH5487 Synthetic H RNAi Description siRNA to inhibit HIST1H4E expression using RNA interference Specificity HIST1H4E siRNA (Human) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol HIST1H4E Alternative Names H4/E; H4FE; Histone H4 Entrez Gene 8367 (Human) SwissProt P62805 (Human) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of human HIST1H4E gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB- Western blot, IH- Immunohistochemistry, -

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

The C-Terminal Region of Histone H4 Recognized by Antinucleosome Th Cells in Identification of a Minimal T Cell Epitope
Identification of a Minimal T Cell Epitope Recognized by Antinucleosome Th Cells in the C-Terminal Region of Histone H4 This information is current as Patrice Decker, Anne Le Moal, Jean-Paul Briand and of October 2, 2021. Sylviane Muller J Immunol 2000; 165:654-662; ; doi: 10.4049/jimmunol.165.2.654 http://www.jimmunol.org/content/165/2/654 Downloaded from References This article cites 34 articles, 13 of which you can access for free at: http://www.jimmunol.org/content/165/2/654.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2000 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Identification of a Minimal T Cell Epitope Recognized by Antinucleosome Th Cells in the C-Terminal Region of Histone H41 Patrice Decker, Anne Le Moal, Jean-Paul Briand, and Sylviane Muller2 Autoreactive T cells responding to systemic autoantigens have been characterized in patients and mice with autoimmune diseases and in healthy individuals. -

Functional Roles of Bromodomain Proteins in Cancer
cancers Review Functional Roles of Bromodomain Proteins in Cancer Samuel P. Boyson 1,2, Cong Gao 3, Kathleen Quinn 2,3, Joseph Boyd 3, Hana Paculova 3 , Seth Frietze 3,4,* and Karen C. Glass 1,2,4,* 1 Department of Pharmaceutical Sciences, Albany College of Pharmacy and Health Sciences, Colchester, VT 05446, USA; [email protected] 2 Department of Pharmacology, Larner College of Medicine, University of Vermont, Burlington, VT 05405, USA; [email protected] 3 Department of Biomedical and Health Sciences, University of Vermont, Burlington, VT 05405, USA; [email protected] (C.G.); [email protected] (J.B.); [email protected] (H.P.) 4 University of Vermont Cancer Center, Burlington, VT 05405, USA * Correspondence: [email protected] (S.F.); [email protected] (K.C.G.) Simple Summary: This review provides an in depth analysis of the role of bromodomain-containing proteins in cancer development. As readers of acetylated lysine on nucleosomal histones, bromod- omain proteins are poised to activate gene expression, and often promote cancer progression. We examined changes in gene expression patterns that are observed in bromodomain-containing proteins and associated with specific cancer types. We also mapped the protein–protein interaction network for the human bromodomain-containing proteins, discuss the cellular roles of these epigenetic regu- lators as part of nine different functional groups, and identify bromodomain-specific mechanisms in cancer development. Lastly, we summarize emerging strategies to target bromodomain proteins in cancer therapy, including those that may be essential for overcoming resistance. Overall, this review provides a timely discussion of the different mechanisms of bromodomain-containing pro- Citation: Boyson, S.P.; Gao, C.; teins in cancer, and an updated assessment of their utility as a therapeutic target for a variety of Quinn, K.; Boyd, J.; Paculova, H.; cancer subtypes. -

Genome-Wide Approach to Identify Risk Factors for Therapy-Related Myeloid Leukemia
Leukemia (2006) 20, 239–246 & 2006 Nature Publishing Group All rights reserved 0887-6924/06 $30.00 www.nature.com/leu ORIGINAL ARTICLE Genome-wide approach to identify risk factors for therapy-related myeloid leukemia A Bogni1, C Cheng2, W Liu2, W Yang1, J Pfeffer1, S Mukatira3, D French1, JR Downing4, C-H Pui4,5,6 and MV Relling1,6 1Department of Pharmaceutical Sciences, The University of Tennessee, Memphis, TN, USA; 2Department of Biostatistics, The University of Tennessee, Memphis, TN, USA; 3Hartwell Center, The University of Tennessee, Memphis, TN, USA; 4Department of Pathology, The University of Tennessee, Memphis, TN, USA; 5Department of Hematology/Oncology St Jude Children’s Research Hospital, The University of Tennessee, Memphis, TN, USA; and 6Colleges of Medicine and Pharmacy, The University of Tennessee, Memphis, TN, USA Using a target gene approach, only a few host genetic risk therapy increases, the importance of identifying host factors for factors for treatment-related myeloid leukemia (t-ML) have been secondary neoplasms increases. defined. Gene expression microarrays allow for a more 4 genome-wide approach to assess possible genetic risk factors Because DNA microarrays interrogate multiple ( 10 000) for t-ML. We assessed gene expression profiles (n ¼ 12 625 genes in one experiment, they allow for a ‘genome-wide’ probe sets) in diagnostic acute lymphoblastic leukemic cells assessment of genes that may predispose to leukemogenesis. from 228 children treated on protocols that included leukemo- DNA microarray analysis of gene expression has been used to genic agents such as etoposide, 13 of whom developed t-ML. identify distinct expression profiles that are characteristic of Expression of 68 probes, corresponding to 63 genes, was different leukemia subtypes.13,14 Studies using this method have significantly related to risk of t-ML. -

Quantitative Proteomics Methods for the Analysis of Histone Post-Translational Modifications
Université de Montréal Quantitative Proteomics Methods for the Analysis of Histone Post-translational Modifications par Nebiyu Ali Abshiru Département de Chimie Faculté des arts et des sciences Thèse présentée à la Faculté des études supérieures et postdoctorales en vue de l’obtention du grade de philosophiae doctor (Ph.D.) en chimie Septembre 2015 ©Nebiyu Ali Abshiru, 2015 i Résumé Les histones sont des protéines nucléaires hautement conservées chez les cellules des eucaryotes. Elles permettent d’organiser et de compacter l’ADN sous la forme de nucléosomes, ceux-ci representant les sous unités de base de la chromatine. Les histones peuvent être modifiées par de nombreuses modifications post-traductionnelles (PTMs) telles que l’acétylation, la méthylation et la phosphorylation. Ces modifications jouent un rôle essentiel dans la réplication de l’ADN, la transcription et l’assemblage de la chromatine. L’abondance de ces modifications peut varier de facon significative lors du developpement des maladies incluant plusieurs types de cancer. Par exemple, la perte totale de la triméthylation sur H4K20 ainsi que l’acétylation sur H4K16 sont des marqueurs tumoraux spécifiques a certains types de cancer chez l’humain. Par conséquent, l’étude de ces modifications et des événements determinant la dynamique des leurs changements d’abondance sont des atouts importants pour mieux comprendre les fonctions cellulaires et moléculaires lors du développement de la maladie. De manière générale, les modifications des histones sont étudiées par des approches biochimiques telles que les immuno-buvardage de type Western ou les méthodes d’immunoprécipitation de la chromatine (ChIP). Cependant, ces approches présentent plusieurs inconvénients telles que le manque de spécificité ou la disponibilité des anticorps, leur coût ou encore la difficulté de les produire et de les valider. -

Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers
Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers writer effector eg. HAT, HMT reader eg. bromodomain eraser eg. HDAC, KDM The writer HAT1: A known H4 lysine 5,12 di-acetyltransferase writer siHAT1 siControl HAT1 H4 K12Ac H4 K5Ac actin Western blot for histone H4 modifications after control and HAT1 siRNA transfections. HAT1: EGF-stimulated immunoprecipitation specific specific - - HAT1 IgG HAT1 IgG - - R α non R α non EGF: + + + - - - WB: HAT1 Immunoprecipitation / WB to measure HAT1 levels +/- Heatmap of gene expression changes of all human histone EGF acetyltransferases +/- EGF and siRNA treatments shows HAT1 expression is EGF-dependent Working model of HAT1 : The oldest “new” histone acetyltransferase EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase Surprise: HAT1 also binds (a few sites) on chromatin HAT1 ChipSeq signal sits on Hist1 locus on Chromosome 6 Read Depth HAT1 bound sites (zoom) HAT1 ChIP-seq peaks cluster at Read Depth histone H4 promoters. Hist1H2BE Hist1H4D Hist1H3D Hist1H4E Is HAT1 a transcription factor for its substrate (H4)? EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase HAT1 is required for S-phase burst of histone H4 mRNA HAT1 Hist1H4B mRNA level 50 Rbap46 45 shCont-3 H4 40 shHAT1-A7 35 shHAT1-B6 30 25 B6 A7 - - 20 15 shHAT1 shHAT1 shControl 10 Gene actin)Expression (versus 5 HAT1 0 0 2 4 6 8 10 actin hours after release from double thymidine block G1 S G2/M HAT1 loss: Life with less histones EGF -

The Role of Histone H2av Variant Replacement and Histone H4 Acetylation in the Establishment of Drosophila Heterochromatin
The role of histone H2Av variant replacement and histone H4 acetylation in the establishment of Drosophila heterochromatin Jyothishmathi Swaminathan, Ellen M. Baxter, and Victor G. Corces1 Department of Biology, Johns Hopkins University, Baltimore, Maryland 21218, USA Activation and repression of transcription in eukaryotes involve changes in the chromatin fiber that can be accomplished by covalent modification of the histone tails or the replacement of the canonical histones with other variants. Here we show that the histone H2A variant of Drosophila melanogaster, H2Av, localizes to the centromeric heterochromatin, and it is recruited to an ectopic heterochromatin site formed by a transgene array. His2Av behaves genetically as a PcG gene and mutations in His2Av suppress position effect variegation (PEV), suggesting that this histone variant is required for euchromatic silencing and heterochromatin formation. His2Av mutants show reduced acetylation of histone H4 at Lys 12, decreased methylation of histone H3 at Lys 9, and a reduction in HP1 recruitment to the centromeric region. H2Av accumulation or histone H4 Lys 12 acetylation is not affected by mutations in Su(var)3-9 or Su(var)2-5. The results suggest an ordered cascade of events leading to the establishment of heterochromatin and requiring the recruitment of the histone H2Av variant followed by H4 Lys 12 acetylation as necessary steps before H3 Lys 9 methylation and HP1 recruitment can take place. [Keywords: Chromatin; silencing; transcription; histone; nucleus] Received September 8, 2004; revised version accepted November 4, 2004. The basic unit of chromatin is the nucleosome, which is guchi et al. 2004). The role of histone variants, and spe- made up of 146 bp of DNA wrapped around a histone cially those of H3 and H2A, in various nuclear processes octamer composed of two molecules each of the histones has been long appreciated (Wolffe and Pruss 1996; Ah- H2A, H2B, H3, and H4. -

Molecular Effects of Isoflavone Supplementation Human Intervention Studies and Quantitative Models for Risk Assessment
Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis committee Promotors Prof. Dr Pieter van ‘t Veer Professor of Nutritional Epidemiology Wageningen University Prof. Dr Evert G. Schouten Emeritus Professor of Epidemiology and Prevention Wageningen University Co-promotors Dr Anouk Geelen Assistant professor, Division of Human Nutrition Wageningen University Dr Lydia A. Afman Assistant professor, Division of Human Nutrition Wageningen University Other members Prof. Dr Jaap Keijer, Wageningen University Dr Hubert P.J.M. Noteborn, Netherlands Food en Consumer Product Safety Authority Prof. Dr Yvonne T. van der Schouw, UMC Utrecht Dr Wendy L. Hall, King’s College London This research was conducted under the auspices of the Graduate School VLAG (Advanced studies in Food Technology, Agrobiotechnology, Nutrition and Health Sciences). Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis submitted in fulfilment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Friday 20 June 2014 at 13.30 p.m. in the Aula. Vera van der Velpen Molecular effects of isoflavone supplementation: Human intervention studies and quantitative models for risk assessment 154 pages PhD thesis, Wageningen University, Wageningen, NL (2014) With references, with summaries in Dutch and English ISBN: 978-94-6173-952-0 ABSTRact Background: Risk assessment can potentially be improved by closely linked experiments in the disciplines of epidemiology and toxicology. -

Genome-Wide Screen of Cell-Cycle Regulators in Normal and Tumor Cells
bioRxiv preprint doi: https://doi.org/10.1101/060350; this version posted June 23, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genome-wide screen of cell-cycle regulators in normal and tumor cells identifies a differential response to nucleosome depletion Maria Sokolova1, Mikko Turunen1, Oliver Mortusewicz3, Teemu Kivioja1, Patrick Herr3, Anna Vähärautio1, Mikael Björklund1, Minna Taipale2, Thomas Helleday3 and Jussi Taipale1,2,* 1Genome-Scale Biology Program, P.O. Box 63, FI-00014 University of Helsinki, Finland. 2Science for Life laboratory, Department of Biosciences and Nutrition, Karolinska Institutet, SE- 141 83 Stockholm, Sweden. 3Science for Life laboratory, Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, S-171 21 Stockholm, Sweden To identify cell cycle regulators that enable cancer cells to replicate DNA and divide in an unrestricted manner, we performed a parallel genome-wide RNAi screen in normal and cancer cell lines. In addition to many shared regulators, we found that tumor and normal cells are differentially sensitive to loss of the histone genes transcriptional regulator CASP8AP2. In cancer cells, loss of CASP8AP2 leads to a failure to synthesize sufficient amount of histones in the S-phase of the cell cycle, resulting in slowing of individual replication forks. Despite this, DNA replication fails to arrest, and tumor cells progress in an elongated S-phase that lasts several days, finally resulting in death of most of the affected cells. -

Histone H4 Lysine 20 Mono-Methylation Directly Facilitates Chromatin Openness and Promotes Transcription of Housekeeping Genes
ARTICLE https://doi.org/10.1038/s41467-021-25051-2 OPEN Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes Muhammad Shoaib 1,8,9, Qinming Chen2,9, Xiangyan Shi 3, Nidhi Nair1, Chinmayi Prasanna 2, Renliang Yang2,4, David Walter1, Klaus S. Frederiksen 5, Hjorleifur Einarsson1, J. Peter Svensson 6, ✉ ✉ ✉ Chuan Fa Liu 2, Karl Ekwall6, Mads Lerdrup 7 , Lars Nordenskiöld 2 & Claus S. Sørensen 1 1234567890():,; Histone lysine methylations have primarily been linked to selective recruitment of reader or effector proteins that subsequently modify chromatin regions and mediate genome functions. Here, we describe a divergent role for histone H4 lysine 20 mono-methylation (H4K20me1) and demonstrate that it directly facilitates chromatin openness and accessibility by disrupting chromatin folding. Thus, accumulation of H4K20me1 demarcates highly accessible chromatin at genes, and this is maintained throughout the cell cycle. In vitro, H4K20me1-containing nucleosomal arrays with nucleosome repeat lengths (NRL) of 187 and 197 are less compact than unmethylated (H4K20me0) or trimethylated (H4K20me3) arrays. Concordantly, and in contrast to trimethylated and unmethylated tails, solid-state NMR data shows that H4K20 mono-methylation changes the H4 conformational state and leads to more dynamic histone H4-tails. Notably, the increased chromatin accessibility mediated by H4K20me1 facilitates gene expression, particularly of housekeeping genes. Altogether, we show how the methy- lation state of a single histone H4 residue operates as a focal point in chromatin structure control. While H4K20me1 directly promotes chromatin openness at highly transcribed genes, it also serves as a stepping-stone for H4K20me3-dependent chromatin compaction. -

SZDB: a Database for Schizophrenia Genetic Research
Schizophrenia Bulletin vol. 43 no. 2 pp. 459–471, 2017 doi:10.1093/schbul/sbw102 Advance Access publication July 22, 2016 SZDB: A Database for Schizophrenia Genetic Research Yong Wu1,2, Yong-Gang Yao1–4, and Xiong-Jian Luo*,1,2,4 1Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences and Yunnan Province, Kunming Institute of Zoology, Kunming, China; 2Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming, China; 3CAS Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences, Shanghai, China 4YGY and XJL are co-corresponding authors who jointly directed this work. *To whom correspondence should be addressed; Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China; tel: +86-871-68125413, fax: +86-871-68125413, e-mail: [email protected] Schizophrenia (SZ) is a debilitating brain disorder with a Introduction complex genetic architecture. Genetic studies, especially Schizophrenia (SZ) is a severe mental disorder charac- recent genome-wide association studies (GWAS), have terized by abnormal perceptions, incoherent or illogi- identified multiple variants (loci) conferring risk to SZ. cal thoughts, and disorganized speech and behavior. It However, how to efficiently extract meaningful biological affects approximately 0.5%–1% of the world populations1 information from bulk genetic findings of SZ remains a and is one of the leading causes of disability worldwide.2–4 major challenge. There is a pressing