2Dre Lichtarge Lab 2006
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Seed Dormancy and the Control of Germination
Review Blackwell Publishing Ltd Tansley review Seed dormancy and the control of germination Author for correspondence: William E. Finch-Savage1 and Gerhard Leubner-Metzger2 G. Leubner-Metzger 1Warwick HRI, Warwick University, Wellesbourne, Warwick CV35 9EF, UK; 2Institute for Biology II, Tel: +49 761 203 2936 Fax: +49 761 203 2612 Botany/Plant Physiology, Albert-Ludwigs-University Freiburg, D-79104 Freiburg i. Br., Germany Email: [email protected] Received: 23 February 2006 Accepted: 8 April 2006 Contents Summary 501 IV. How is nondeep physiological seed dormancy regulated by the environment? Ecophysiology and modelling 514 I. Introduction 502 V. Conclusions and perspectives 518 II. What is dormancy and how is it related to germination? 502 Acknowledgements 519 III. How is nondeep physiological dormancy regulated within the seed at the molecular level? 509 References 519 Supplementary material 523 Summary Seed dormancy is an innate seed property that defines the environmental con- ditions in which the seed is able to germinate. It is determined by genetics with a substantial environmental influence which is mediated, at least in part, by the plant hormones abscisic acid and gibberellins. Not only is the dormancy status influenced by the seed maturation environment, it is also continuously changing with time following shedding in a manner determined by the ambient environ- ment. As dormancy is present throughout the higher plants in all major climatic regions, adaptation has resulted in divergent responses to the environment. Through this adaptation, germination is timed to avoid unfavourable weather for subsequent plant establishment and reproductive growth. In this review, we present an integrated view of the evolution, molecular genetics, physiology, biochemistry, ecology and modelling of seed dormancy mechanisms and their control of germination. -

A Synopsis of the South American Lepidium (Brassicaceae)
DARWINIANA 48(2): 141-167. 2010 ISSN 0011-6793 A SYNOPSIS OF THE SOUTH AMERICAN LEPIDIUM (BRASSICACEAE) Ihsan A. Al-Shehbaz Missouri Botanical Garden, P.O. Box 299, St. Louis, Missouri 63166-0299, U.S.A.; [email protected] Abstract. Al-Shehbaz, I. A. 2010. A synopsis of the South American Lepidium (Brassicaceae). Darwiniana 48(2): 141-167. Fifty native and 12 naturalized species of Lepidium grow in South America, and a key to all 62 spe- cies is presented. Three new species from Argentina (L. hickenii, L. pedersenii, and L. santacruzensis) and two from Peru (L. cuzcoensis and L. werffii) are described, illustrated, and their relationships to nearest relatives are discussed. The new combination Lepidium crassius is proposed. Furthermore, 19 taxa (Coronopus didymus var. macrocarpus, C. didymus var. procumbens, L. abrotanifolium var. stein- mannii, L. affine, L. argentinum, L. auriculatum, L. bonariense var. pseudovirginicum, L. calycinum var. integrifolium, L. costaricense, L. cumingianum var. orbiculatum, L. cumingianum subsp. bertero- anum, L. depressum, L. myrianthum, L. neglectum, L. parodii, L. pubescens var. fallax, L. subvagina- tum, L. virginicum subsp. centrali-americanum, Thlaspi campestre) are lectotypified and 17 others (Coronopus leptocarpus, C. leptocarpus var. microcarpus, L. boelckii, L. bonariense var. gayi, L. bonariense var. pseudovirginicum, L. brevicaule, L. calycinum, L. danielsii, L. demissum, L. kalenbor- nii, L. morrisonii, L. peruvianum, L. philippianum var. boliviense, L. raimondii, L. scabrifructum, L. spicatum var. caylx-persistente, L. subvaginatum) are reduced to synonymy. L. depressum and L. rah- meri are new records for Argentina. Keywords. Brassicaceae, Lepidium, South America, taxonomy. Resumen. Al-Shehbaz, I. -

London Rocket Tech Bulletin – ND
4/6/2020 London Rocket London Rocket Sisymbrium irio L. Family: Brassicaceae. Names: Sisymbrium was the Greek name of a fragrant herb. London Rocket. Summary: An erect, annual, many branched plant, with deeply lobed leaves that does not form a rosette. It has clusters of small, 4-petalled, yellow flowers in late winter to spring on the tops of stems that form long (25-110 mm), narrow seed pods that may be slightly curved. Description: Cotyledons: Two. Club shaped, Tip rounded. Sides convex. Base tapered. Surface hairless. Petiole longer than the blade. First Leaves: Club shaped, paired. The first pair have rounded tips and smooth edges. The second pair have pointed tips and toothed edges. Hairless or a few hairs. Leaves: Alternate. Does not form a rosette. Stipules - None. Petiole - On lower leaves. Blade - 30-160 mm long x 13-70 mm wide, triangular in outline, deeply lobed or serrated or toothed (usually 2-6 pairs), lobes are usually toothed, end lobe is pointed and larger than the side lobes. The side lobes usually point towards the base of the leaf. Tip pointed. Smooth and hairless or a few scattered hairs. Stem leaves - Alternate. Similar to rosette leaves but not as lobed or unlobed, sometimes arrow shaped. Hairless or small hairs. Stems: Slender, erect, round, up to 1000 mm tall. Often with slender, curved, simple hairs near the base, usually hairless near the top. Usually much branched from the base with spreading stems. Flower head: www.herbiguide.com.au/Descriptions/hg_London_Rocket.htm 1/8 4/6/2020 London Rocket Flowers are in clusters at the top of the stem which then elongates as the fruits mature underneath. -

Plants of Hot Springs Valley and Grover Hot Springs State Park Alpine County, California
Plants of Hot Springs Valley and Grover Hot Springs State Park Alpine County, California Compiled by Tim Messick and Ellen Dean This is a checklist of vascular plants that occur in Hot Springs Valley, including most of Grover Hot Springs State Park, in Alpine County, California. Approximately 310 taxa (distinct species, subspecies, and varieties) have been found in this area. How to Use this List Plants are listed alphabetically, by family, within major groups, according to their scientific names. This is standard practice for plant lists, but isn’t the most user-friendly for people who haven’t made a study of plant taxonomy. Identifying species in some of the larger families (e.g. the Sunflowers, Grasses, and Sedges) can become very technical, requiring examination of many plant characteristics under high magnification. But not to despair—many genera and even species of plants in this list become easy to recognize in the field with only a modest level of study or help from knowledgeable friends. Persistence will be rewarded with wonder at the diversity of plant life around us. Those wishing to pursue plant identification a bit further are encouraged to explore books on plants of the Sierra Nevada, and visit CalPhotos (calphotos.berkeley.edu), the Jepson eFlora (ucjeps.berkeley.edu/eflora), and CalFlora (www.calflora.org). The California Native Plant Society (www.cnps.org) promotes conservation of plants and their habitats throughout California and is a great resource for learning and for connecting with other native plant enthusiasts. The Nevada Native Plant Society nvnps.org( ) provides a similar focus on native plants of Nevada. -
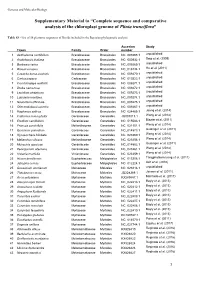
Complete Sequence and Comparative Analysis of the Chloroplast Genome of Plinia Trunciflora”
Genetics and Molecular Biology Supplementary Material to “Complete sequence and comparative analysis of the chloroplast genome of Plinia trunciflora” Table S3 - List of 56 plastome sequences of Rosids included in the Bayesian phylogenetic analysis. Accesion Study Taxon Family Order number 1 Aethionema cordifolium Brassicaceae Brassicales NC_009265.1 unpublished 2 Arabidopsis thaliana Brassicaceae Brassicales NC_000932.1 Sato et al. (1999) 3 Barbarea verna Brassicaceae Brassicales NC_009269.1 unpublished 4 Brassica napus Brassicaceae Brassicales NC_016734.1 Hu et al. (2011) 5 Capsella bursa-pastoris Brassicaceae Brassicales NC_009270.1 unpublished 6 Carica papaya Caricaceae Brassicales NC_010323.1 unpublished 7 Crucihimalaya wallichii Brassicaceae Brassicales NC_009271.1 unpublished 8 Draba nemorosa Brassicaceae Brassicales NC_009272.1 unpublished 9 Lepidium virginicum Brassicaceae Brassicales NC_009273.1 unpublished 10 Lobularia maritima Brassicaceae Brassicales NC_009274.1 unpublished 11 Nasturtium officinale Brassicaceae Brassicales NC_009275.1 unpublished 12 Olimarabidopsis pumila Brassicaceae Brassicales NC_009267.1 unpublished 13 Raphanus sativus Brassicaceae Brassicales NC_024469.1 Jeong et al. (2014) 14 California macrophylla Geraniaceae Geraniales JQ031013.1 Weng et al. (2014) 15 Erodium carvifolium Geraniaceae Geraniales NC_015083.1 Blazier et al. (2011) 16 Francoa sonchifolia Melianthaceae Geraniales NC_021101.1 Weng et al. (2014) 17 Geranium palmatum Geraniaceae Geraniales NC_014573.1 Guisinger et al. (2011) 18 Hypseocharis bilobate Geraniaceae Geraniales NC_023260.1 Weng et al. (2014) 19 Melianthus villosus Melianthaceae Geraniales NC_023256.1 Weng et al. (2014) 20 Monsonia speciose Geraniaceae Geraniales NC_014582.1 Guisinger et al. (2011) 21 Pelargonium alternans Geraniaceae Geraniales NC_023261.1 Weng et al. (2014) 22 Viviania marifolia Vivianiaceae Geraniales NC_023259.1 Weng et al. (2014) 23 Hevea brasiliensis Euphorbiaceae Malpighiales NC_015308.1 Tangphatsornruang et al. -

Host Choice in Rotylenchulus Species
Available online at www.ijpab.com Rathore Int. J. Pure App. Biosci. 6 (5): 346-354 (2018) ISSN: 2320 – 7051 DOI: http://dx.doi.org/10.18782/2320-7051.6878 ISSN: 2320 – 7051 Int. J. Pure App. Biosci. 6 (5): 346-354 (2018) Research Article Host Choice in Rotylenchulus Species Y. S. Rathore* Principal Scientist (Retd.), Indian Institute of Pulses Research, Kanpur-208 024 (U.P.) India *Corresponding Author E-mail: [email protected] Received: 12.09.2018 | Revised: 9.10.2018 | Accepted: 16.10.2018 ABSTRACT The reniformis nematodes of the genus Rotylenchulus (Haplolaimidae: Nematoda) are sedentary semi-endoparasites of numerous crops. There are ten species out of which R. reniformis and R. parvus are important, and three species (R. amanictus, R. clavicadatus, R. leptus) are monophagous: two on monocots and one on Rosids. In general, Rotylenchulus species are capable of feeding from very primitive Magnoliids to plants of advanced category. Preference was distinctly observed towards the plants in Rosids (42.779%) followed by monocots (23.949%) and Asterids (21.755%). The SAI values were also higher for these groups of plants. The study on lineages further revealed intimate affinity to febids (25.594%), followed by commelinids (18.647%), malvids (16.088%), lamiids (11.883%), and campanulids (9.141%). Poales contribution within commelinids was 65.353%. Maximum affinity of Rotylenchulus species was observed by their association with plants from families Poaceae (7), followed by Fabaceae (6), Malvaceae (6), Asteraceae (4), Oleaceae (4), Soanaceae (4) and so on. Key words: Agiosperms, Gymnosperms, APG IV system, Reniform nemtodes, Monocots, Rosids, Asterids INTRODUCTION number of crops, whereas the other eight Plant parasitic nematodes pose a great species are of limited importance. -

Checklist of the Vascular Plants of San Diego County 5Th Edition
cHeckliSt of tHe vaScUlaR PlaNtS of SaN DieGo coUNty 5th edition Pinus torreyana subsp. torreyana Downingia concolor var. brevior Thermopsis californica var. semota Pogogyne abramsii Hulsea californica Cylindropuntia fosbergii Dudleya brevifolia Chorizanthe orcuttiana Astragalus deanei by Jon P. Rebman and Michael G. Simpson San Diego Natural History Museum and San Diego State University examples of checklist taxa: SPecieS SPecieS iNfRaSPecieS iNfRaSPecieS NaMe aUtHoR RaNk & NaMe aUtHoR Eriodictyon trichocalyx A. Heller var. lanatum (Brand) Jepson {SD 135251} [E. t. subsp. l. (Brand) Munz] Hairy yerba Santa SyNoNyM SyMBol foR NoN-NATIVE, NATURaliZeD PlaNt *Erodium cicutarium (L.) Aiton {SD 122398} red-Stem Filaree/StorkSbill HeRBaRiUM SPeciMeN coMMoN DocUMeNTATION NaMe SyMBol foR PlaNt Not liSteD iN THE JEPSON MANUAL †Rhus aromatica Aiton var. simplicifolia (Greene) Conquist {SD 118139} Single-leaF SkunkbruSH SyMBol foR StRict eNDeMic TO SaN DieGo coUNty §§Dudleya brevifolia (Moran) Moran {SD 130030} SHort-leaF dudleya [D. blochmaniae (Eastw.) Moran subsp. brevifolia Moran] 1B.1 S1.1 G2t1 ce SyMBol foR NeaR eNDeMic TO SaN DieGo coUNty §Nolina interrata Gentry {SD 79876} deHeSa nolina 1B.1 S2 G2 ce eNviRoNMeNTAL liStiNG SyMBol foR MiSiDeNtifieD PlaNt, Not occURRiNG iN coUNty (Note: this symbol used in appendix 1 only.) ?Cirsium brevistylum Cronq. indian tHiStle i checklist of the vascular plants of san Diego county 5th edition by Jon p. rebman and Michael g. simpson san Diego natural history Museum and san Diego state university publication of: san Diego natural history Museum san Diego, california ii Copyright © 2014 by Jon P. Rebman and Michael G. Simpson Fifth edition 2014. isBn 0-918969-08-5 Copyright © 2006 by Jon P. -

Confused by Crucifers?--A Mustard Identification Workshop Tim Miller, Extension Weed Scientist, WSU--Mount Vernon Washington State Weed Conference November 4, 1998
Confused by Crucifers?--A Mustard Identification Workshop Tim Miller, Extension Weed Scientist, WSU--Mount Vernon Washington State Weed Conference November 4, 1998 WHAT DOES “MUSTARD” MEAN? Mustard is the common household and food industry term for the yellow or brown table condiment so often eaten on hot dogs, as well as the agricultural term for the plant that produces the seeds from which that condiment is made. It is also the common name of the plant family known as Brassicaceae (also called Cruciferae). Any species within that family can be commonly (but loosely) called a “mustard” or a “crucifer.” Our word “mustard” comes from the Latin mustum and Old French mustarde, meaning “must” (a term for the fruit and skins of crushed grapes), perhaps a reference to the crushing of mustard seeds necessary to produce the pungent yellow powder. Botanically speaking, what makes a mustard a mustard? Three major characteristics separate the mustard family from all other plant families: 1. The stamens are tetradynamous, meaning there are four long stamens and two short stamens in each flower (six in total). 2. Each flower bears four petals that form a cross, hence the alternate family name “Cruciferae,” from the Latin cruciform which means “cross-shaped.” 3. The seed pods each have a thin, translucent inner membrane, the replum, that separates the two chambers of the pod, and to which the seeds are attached. Remember: although an unknown plant specimen may possess one or even two of these characteristics, that does not necessarily mean you are looking at a mustard species. It is the combination of all three characteristics that defines the mustard family. -
1 Checklist of Plants: Balcones Canyonlands
CHECKLIST OF PLANTS: BALCONES CANYONLANDS NATIONAL WILDLIFE REFUGE September 24, 2008 Note : Species marked with an asterisk (*) are endemic to Texas (most often confined to just Central Texas or the Edwards Plateau). (I) indicates introduced (non-native) species. Species in square [brackets] are found on lands immediately adjacent to the Refuge and might be expected within the Refuge boundary. SCIENTIFIC NAME COMMON NAME ACANTHACEAE ACANTHUS FAMILY Dyschoriste linearis Narrowleaf dyschoriste, Snake herb Justicia americana American water-willow *Ruellia drummondiana *Drummond wild-petunia Ruellia humilis Low wild-petunia Ruellia metziae Common wild-petunia Ruellia nudiflora var. nudiflora Common wild-petunia Siphonoglossa pilosella Tube-tongue ACERACEAE MAPLE FAMILY Acer negundo Box-elder AGAVACEAE AGAVE FAMILY *Nolina lindheimeriana *Devil's shoestring Nolina texana Sacahuista Yucca arkansana Arkansas yucca Yucca constricta Buckley yucca *Yucca rupicola *Twist-leaf yucca Yucca treculeana Trecul yucca AMARANTHACEAE AMARANTH FAMILY Alternanthera caracasana Mat chaff-flower Amaranthus albus White amaranth Amaranthus palmeri Palmer's amaranth Amaranthus retroflexus Redroot pigweed Amaranthus sp. Amaranth Froelichia gracilis Snake-cotton ANACARDIACEAE SUMAC FAMILY Rhus lanceolata Flameleaf sumac Rhus trilobata var. trilobata Fragrant sumac, skunkbush Rhus virens Evergreen sumac Toxicodendron radicans Poison ivy (prob. 2 vars.) APIACEAE (= Umbelliferae) CARROT FAMILY Bifora americana Prairie bishop Bowlesia incana Hoary bowlesia Centella erecta -
Vascular Plant List Deception Pass Deception Pass, Island and Skagit Counties, WA
Vascular Plant List Deception Pass Deception Pass, Island and Skagit Counties, WA. List covers plants found in Deception Pass State Park, including areas on both sides of Deception Pass. WNPS list over several years, through 2000. 238 spp. * - Introduced Scientific Name Common Name Family Name Family Common Color Ref Acer Glabrum var.douglasii Douglas Maple Aceraceae Maple Family White Acer macrophyllum Big-leaf maple Aceraceae White Daucus pusillus American carrot Apiaceae Parsley Family White Glehnia leiocarpa American glehnia Apiaceae White Heracleum lanatum Cow parsnip Apiaceae White Lomatium utriculatum Spring gold Apiaceae Yellow Oenanthe sarmentosa Water parsley Apiaceae White Osmorhiza chilensis Mountain sweet-cicely Apiaceae Grn/White Sanicula bipinnatifida Purple sanicle Apiaceae Purple Sanicula crassicaulis Pacific sanicle Apiaceae Yellow Sium suave Water-parsnip Apiaceae White Ilex aquifolium* English holly Aquifoliaceae Holly Family White Lysichiton americanum Skunk cabbage Araceae Yellow Hedera helix* English ivy Araliaceae White Achillea millifolium Yarrow Asteraceae White Adenocaulon bicolor Pathfinder Asteraceae White Ambrosia chamissonis Silver bursage Asteraceae Yellow Anaphalis margaritacea Pearly everlasting Asteraceae White Arctium minus* Common burdock Asteraceae Pink Artemesia campestris Northern wormwood Asteraceae Yellow Bellis perennis* English daisy Asteraceae White/yel Chrysanthemum leucanthemum* Ox-eye daisy Asteraceae White/yel Cirsium arvense* Canada thistle Asteraceae Pink Cirsium brevistylum Indian thistle -

Virginia Pepperweed (Lepidium Virginicum)
Virginia pepperweed (Lepidium virginicum) For definitions of botanical terms, visit en.wikipedia.org/wiki/Glossary_of_botanical_terms. Virginia pepperweed begins as a low-growing basal rosette of deeply lobed leaves. Arising from the rosette on branching stems are elongated racemes of tiny whitish-yellow flowers. They have the appearance of thin, greenish-white bottlebrushes. Leaves are linear to lanceolate and toothed. Seeds develop along the stem and vary in shape but most commonly resemble a flat, lentil-shaped disk. Virginia pepperweed flowers are hermaphroditic (have both male and female organs) and are pollinated by a variety of insects. Bees love Virginia pepperweed. It is also the host plant for the checkered white (Pontia protodice) and great Southern white (Ascia monuste) butterflies. Photo by Forest and Kim Starr (Creative Commons license) Virginia pepperweed is a member of the mustard family and is edible to humans. The young leaves, which contain protein and vitamins A and C, can be added to salads or sauteed; the seed pods can be used as a substitute for black pepper. (Doubtful? Pop a few clean seeds in your mouth and you will know immediately why this Florida native is sometimes called poor man’s pepper.) Family: Brassicaceae (Mustard family) Native range: Throughout Florida except Nassau and Glades counties To see where natural populations of Virginia pepperweed have been vouchered, visit www.florida.plantatlas.usf.edu. Hardiness: Zones 7–11 Soil: Dry, well-drained, sandy to rich soils Exposure: Full sun Growth habit: 6–20” tall Garden tips: If eating Virginia pepperweed doesn’t appeal to you, the dried seed stems can also be used in flower arrangements. -

Western Washington Plant List By
The NatureMapping Program Western Washington Plant List Revised: 5/21/2011 (1) Non- native, (2) ID Scientific Name Common Name Plant Family Invasive 2 Acer circinatum Vine maple Aceraceae 763 Acer glabrum Douglas maple Aceraceae 3 Acer macrophyllum Big-leaf maple Aceraceae 470 Acer platinoides* Norway maple Aceraceae 1 484 Acer pseudoplatanus* Sycamore maple Aceraceae 1 4 Acer saccharum* Sugar maple Aceraceae 1 727 Viburnum edule Squashberry Adoxaceae 19 Alisma plantago-aquatica American waterplantain Alismataceae 349 Sagittaria latifolia Broadleaf arrowhead Alismataceae 24 Amaranthus powellii* Powell's amaranth Amaranthaceae 1 255 Narcissus pseudonarcissus* Daffodil Amaryllidaceae 1 327 Rhus diversiloba Poison oak Anacardiaceae 29 Angelica arguta Sharp-tooth angelica Apiaceae 30 Angelica genuflexa Kneeling angelica Apiaceae 31 Angelica lucida Sea-watch Apiaceae 34 Anthriscus scandicina* Chervil Apiaceae 1 96 Conium maculatum* Poison hemlock Apiaceae 1 113 Daucus carota* Queen Anne's lace Apiaceae 1 167 Heracleum lanatum Cow parsnip Apiaceae 766 Heracleum mantegazzianum Giant hogweed Apiaceae 1 175 Hydrocotyle ranunculoides Marsh penny-wort Apiaceae 204 Lilaeopsis occidentalis Lilaeopsis Apiaceae 261 Oenanthe sarmentosa Water parsley Apiaceae 474 Osmorhiza chilensis Sweet-cicely Apiaceae 264 Osmorhiza occidentalis Western sweet-cicely Apiaceae 359 Sanicula crassicaulis Pacific sanicle Apiaceae 492 Sanicula graveolens Northern sanicle Apiaceae 450 Vinca major* Periwinkle Apocynaceae 1 180 Ilex aquifolium* English holly Aquifoliaceae 2