Tyrosine Kinase Inhibition Increases the Cell Surface Localization of FLT3-ITD and Enhances FLT3-Directed Immunotherapy of Acute Myeloid Leukemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Promoter Janus Kinase 3 Proximal Characterization and Analysis Of
The Journal of Immunology Characterization and Analysis of the Proximal Janus Kinase 3 Promoter1 Martin Aringer,2*† Sigrun R. Hofmann,2* David M. Frucht,* Min Chen,* Michael Centola,* Akio Morinobu,* Roberta Visconti,* Daniel L. Kastner,* Josef S. Smolen,† and John J. O’Shea3* Janus kinase 3 (Jak3) is a nonreceptor tyrosine kinase essential for signaling via cytokine receptors that comprise the common ␥-chain (␥c), i.e., the receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. Jak3 is preferentially expressed in hemopoietic cells and is up-regulated upon cell differentiation and activation. Despite the importance of Jak3 in lymphoid development and immune function, the mechanisms that govern its expression have not been defined. To gain insight into this issue, we set out to characterize the Jak3 promoter. The 5-untranslated region of the Jak3 gene is interrupted by a 3515-bp intron. Upstream of this intron and the transcription initiation site, we identified an ϳ1-kb segment that exhibited lymphoid-specific promoter activity and was responsive to TCR signals. Truncation of this fragment revealed that core promoter activity resided in a 267-bp fragment that contains putative Sp-1, AP-1, Ets, Stat, and other binding sites. Mutation of the AP-1 sites significantly diminished, whereas mutation of the Ets sites abolished, the inducibility of the promoter construct. Chromatin immunoprecipitation assays showed that histone acetylation correlates with mRNA expression and that Ets-1/2 binds this region. Thus, transcription factors that bind these sites, especially Ets family members, are likely to be important regulators of Jak3 expression. -

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

CD137 Microbead Kit CD137 Microbead + Cells
CD137 MicroBead Kit human Order no. 130-093-476 Contents 1.2 Background information 1. Description The activation-induced antigen CD137 (4-1BB) is a 30 kDa glycoprotein of the tumor necrosis factor (TNF) receptor 1.1 Principle of the MACS® Separation + + superfamily. It is mainly expressed on activated CD4 and CD8 1.2 Background information T cells, activated B cells, and natural killer cells, but can also be 1.3 Applications found on resting monocytes and dendritic cells. As a costimulatory molecule, CD137 is involved in the activation 1.4 Reagent and instrument requirements and survival of CD4, CD8, and NK cells. Its engagement enhances 2. Protocol expansion of T cells and activates them to secrete cytokines. CD137 has been described to be a suitable marker for antigen- 2.1 Sample preparation specific activation of human CD8+ T cells, as CD137 is not expressed 2.2 Magnetic labeling on resting CD8+ T cells and its expression is reliably induced after 2.3 Magnetic separation 24 hours of stimulation.¹,² 3. Example of a separation using the CD137 MicroBead Kit 1.3 Applications 4. References ● Enrichment of CD137+ T cells for phenotypical and functional 5. Appendix characterization. ● Enrichment of activated antigen-specific T cells after antigen- Warnings specific stimulation. Reagents contain sodium azide. Under acidic conditions sodium 1.4 Reagent and instrument requirements azide yields hydrazoic acid, which is extremely toxic. Azide ● compounds should be diluted with running water before discarding. Buffer: Prepare a solution containing phosphate-buffered These precautions are recommended to avoid deposits in plumbing saline (PBS), pH 7.2, 0.5% bovine serum albumin (BSA), where explosive conditions may develop. -

Ret Oncogene and Thyroid Carcinoma
ndrom Sy es tic & e G n e e n G e f T o Elisei et al., J Genet Syndr Gene Ther 2014, 5:1 Journal of Genetic Syndromes h l e a r n a DOI: 10.4172/2157-7412.1000214 r p u y o J & Gene Therapy ISSN: 2157-7412 Review Article Open Access Ret Oncogene and Thyroid Carcinoma Elisei R, Molinaro E, Agate L, Bottici V, Viola D, Biagini A, Matrone A, Tacito A, Ciampi R, Vivaldi A and Romei C* Endocrine Unit, Department of Clinical and Experimental Medicine, University of Pisa, Italy Abstract Thyroid cancer is a malignant neoplasm that originates from follicular or parafollicular thyroid cells and is categorized as papillary (PTC), follicular (FTC), anaplastic (ATC) or medullary thyroid carcinoma (MTC). The alteration of the Rearranged during trasfection (RET) (proto-oncogene, a gene coding for a tyrosine-kinase receptor involved in the control of cell differentiation and proliferation, has been found to cause PTC and MTC. In particular, RET/PTC rearrangements and RET point mutations are related to PTC and MTC, respectively. Although RET/PTC rearrangements have been identified in both spontaneous and radiation-induced PTC, they occur more frequently in radiation-associated tumors. RET/PTC rearrangements have also been reported in follicular adenomas. Although controversial, correlations between RET/PTC rearrangements, especially RET/PTC3, and a more aggressive phenotype and a more advanced stage have been identified. Germline point mutations in the RET proto-oncogene are associated with nearly all cases of hereditary MTC, and a strict correlation between genotype and phenotype has been demonstrated. -

A Gain-Of-Function P53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia Through the Pluripotency Factor FOXH1
Published OnlineFirst May 8, 2019; DOI: 10.1158/2159-8290.CD-18-1391 RESEARCH ARTICLE A Gain-of-Function p53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia through the Pluripotency Factor FOXH1 Evangelia Loizou1,2, Ana Banito1, Geulah Livshits1, Yu-Jui Ho1, Richard P. Koche3, Francisco J. Sánchez-Rivera1, Allison Mayle1, Chi-Chao Chen1, Savvas Kinalis4, Frederik O. Bagger4,5, Edward R. Kastenhuber1,6, Benjamin H. Durham7, and Scott W. Lowe1,8 Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Published OnlineFirst May 8, 2019; DOI: 10.1158/2159-8290.CD-18-1391 ABSTRACT Mutations in the TP53 tumor suppressor gene are common in many cancer types, including the acute myeloid leukemia (AML) subtype known as complex karyotype AML (CK-AML). Here, we identify a gain-of-function (GOF) Trp53 mutation that accelerates CK-AML initiation beyond p53 loss and, surprisingly, is required for disease maintenance. The Trp53 R172H muta- tion (TP53 R175H in humans) exhibits a neomorphic function by promoting aberrant self-renewal in leu- kemic cells, a phenotype that is present in hematopoietic stem and progenitor cells (HSPC) even prior to their transformation. We identify FOXH1 as a critical mediator of mutant p53 function that binds to and regulates stem cell–associated genes and transcriptional programs. Our results identify a context where mutant p53 acts as a bona fi de oncogene that contributes to the pathogenesis of CK-AML and suggests a common biological theme for TP53 GOF in cancer. SIGNIFICANCE: Our study demonstrates how a GOF p53 mutant can hijack an embryonic transcrip- tion factor to promote aberrant self-renewal. -

Human B Cell Isolation Product Selection Diagram
Human B Cell Isolation Product Selection Explore the infographic below to find the correct human B cell isolation product for your application. 1. Your Starting Sample Whole Peripheral Blood/Buffy Coat PBMCs/Leukapheresis Pack 2. Cell Separation Platform Immunodensity Cell Separation Immunomagnetic Cell Separation Immunomagnetic Cell Separation 3. Product Line RosetteSep™ EasySep™ EasySep™ Sequential Selection Negative Selection Negative Selection Positive Selection Negative Selection Positive Selection 4. Selection Method (Positive + Negative) iRosetteSep™ HLA iEasySep™ Direct HLA i, iiEasySep™ HLA iEasySep™ HLA B Cell viEasySep™ Human CD19 EasySep™ Human IgG+ B Cell Enrichment Cocktail B Cell Isolation Kit Chimerism Whole Blood Enrichment Kit Positive Selection Kit II Memory B Cell Isolation (15064HLA)1, 2, 3 (89684) / EasySep™ Direct B Cell Positive Selection (19054HLA)1, 2 (17854)1, 2 Kit (17868)1 (optional), 2' HLA Crossmatch B Cell Kit (17886)1, 2 Isolation Kit (19684 - 1, 2 RosetteSep™ Human available in the US only) iiiEasySep™ Human B Cell viEasySep™ Release EasySep™ Human Memory 5. Cell Isolation Kits B Cell Enrichment Cocktail i, iiEasySep™ HLA Enrichment Kit Human CD19 Positive B Cell Isolation Kit 1, 2, 3 1, 2 1, 2 1 (optional), 2 Catalog #s shown in ( ) (15024) Chimerism Whole Blood (19054) Selection Kit (17754) (17864) EasySep™ Direct Human CD19 Positive Selection B-CLL Cell Isolation Kit Kit (17874)1, 2 1, 2, 3, 4 *RosetteSep™ Human (19664) iiiEasySep™ Human B Cell EasySep™ Human CD138 Multiple Myeloma Cell Isolation Kit -

FLT3 Inhibitors in Acute Myeloid Leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2*
Wu et al. Journal of Hematology & Oncology (2018) 11:133 https://doi.org/10.1186/s13045-018-0675-4 REVIEW Open Access FLT3 inhibitors in acute myeloid leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2* Abstract FLT3 mutations are one of the most common findings in acute myeloid leukemia (AML). FLT3 inhibitors have been in active clinical development. Midostaurin as the first-in-class FLT3 inhibitor has been approved for treatment of patients with FLT3-mutated AML. In this review, we summarized the preclinical and clinical studies on new FLT3 inhibitors, including sorafenib, lestaurtinib, sunitinib, tandutinib, quizartinib, midostaurin, gilteritinib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG 925, TTT-3002, and FF-10101. New generation FLT3 inhibitors and combination therapies may overcome resistance to first-generation agents. Keywords: FMS-like tyrosine kinase 3 inhibitors, Acute myeloid leukemia, Midostaurin, FLT3 Introduction RAS, MEK, and PI3K/AKT pathways [10], and ultim- Acute myeloid leukemia (AML) remains a highly resist- ately causes suppression of apoptosis and differentiation ant disease to conventional chemotherapy, with a me- of leukemic cells, including dysregulation of leukemic dian survival of only 4 months for relapsed and/or cell proliferation [11]. refractory disease [1]. Molecular profiling by PCR and Multiple FLT3 inhibitors are in clinical trials for treat- next-generation sequencing has revealed a variety of re- ing patients with FLT3/ITD-mutated AML. In this re- current gene mutations [2–4]. New agents are rapidly view, we summarized the preclinical and clinical studies emerging as targeted therapy for high-risk AML [5, 6]. on new FLT3 inhibitors, including sorafenib, lestaurtinib, In 1996, FMS-like tyrosine kinase 3/internal tandem du- sunitinib, tandutinib, quizartinib, midostaurin, gilteriti- plication (FLT3/ITD) was first recognized as a frequently nib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG mutated gene in AML [7]. -

Wnt-Independent and Wnt-Dependent Effects of APC Loss on the Chemotherapeutic Response
International Journal of Molecular Sciences Review Wnt-Independent and Wnt-Dependent Effects of APC Loss on the Chemotherapeutic Response Casey D. Stefanski 1,2 and Jenifer R. Prosperi 1,2,3,* 1 Department of Biological Sciences, University of Notre Dame, Notre Dame, IN 46617, USA; [email protected] 2 Mike and Josie Harper Cancer Research Institute, South Bend, IN 46617, USA 3 Department of Biochemistry and Molecular Biology, Indiana University School of Medicine-South Bend, South Bend, IN 46617, USA * Correspondence: [email protected]; Tel.: +1-574-631-4002 Received: 30 September 2020; Accepted: 20 October 2020; Published: 22 October 2020 Abstract: Resistance to chemotherapy occurs through mechanisms within the epithelial tumor cells or through interactions with components of the tumor microenvironment (TME). Chemoresistance and the development of recurrent tumors are two of the leading factors of cancer-related deaths. The Adenomatous Polyposis Coli (APC) tumor suppressor is lost in many different cancers, including colorectal, breast, and prostate cancer, and its loss correlates with a decreased overall survival in cancer patients. While APC is commonly known for its role as a negative regulator of the WNT pathway, APC has numerous binding partners and functional roles. Through APC’s interactions with DNA repair proteins, DNA replication proteins, tubulin, and other components, recent evidence has shown that APC regulates the chemotherapy response in cancer cells. In this review article, we provide an overview of some of the cellular processes in which APC participates and how they impact chemoresistance through both epithelial- and TME-derived mechanisms. Keywords: adenomatous polyposis coli; chemoresistance; WNT signaling 1. -

Insulin–Insr Signaling Drives Multipotent Progenitor Differentiation Toward Lymphoid Lineages
Article Insulin–InsR signaling drives multipotent progenitor differentiation toward lymphoid lineages Pengyan Xia,1* Shuo Wang,1* Ying Du,1 Guanling Huang,1,2 Takashi Satoh,3 Shizuo Akira,3 and Zusen Fan1 1Key Laboratory of Infection and Immunity of CAS, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China 2University of Chinese Academy of Sciences, Beijing 100049, China 3Department of Host Defense, Research Institute for Microbial Diseases (RIMD), Osaka University, Suita, Osaka 565-0871, Japan The lineage commitment of HSCs generates balanced myeloid and lymphoid populations in hematopoiesis. However, the un- derlying mechanisms that control this process remain largely unknown. Here, we show that insulin–insulin receptor (InsR) signaling is required for lineage commitment of multipotent progenitors (MPPs). Deletion of Insr in murine bone marrow causes skewed differentiation of MPPs to myeloid cells. mTOR acts as a downstream effector that modulates MPP differentiation. mTOR activates Stat3 by phosphorylation at serine 727 under insulin stimulation, which binds to the promoter of Ikaros, lead- ing to its transcription priming. Our findings reveal that the insulin–InsR signaling drives MPP differentiation into lymphoid lineages in early lymphopoiesis, which is essential for maintaining a balanced immune system for an individual organism. Hematopoiesis is the process of producing all compo- Insulin, as the primary anabolic hormone, modulates a nents of the blood system from hematopoietic stem cells variety of physiological processes, including growth, differ- (HSCs; Naik et al., 2013; Mendelson and Frenette, 2014; entiation, apoptosis, and synthesis and breakdown of lipid, Walter et al., 2015). HSCs are quiescent, self-renewable pro- protein, and glucose (Samuel and Shulman, 2012). -

Transient Exposure to Quizartinib Mediates Sustained Inhibition of FLT3 Signaling While Specifically Inducing Apoptosis in FLT3-Activated Leukemia Cells
Published OnlineFirst February 14, 2013; DOI: 10.1158/1535-7163.MCT-12-0305 Molecular Cancer Cancer Therapeutics Insights Therapeutics Transient Exposure to Quizartinib Mediates Sustained Inhibition of FLT3 Signaling while Specifically Inducing Apoptosis in FLT3-Activated Leukemia Cells Ruwanthi N. Gunawardane, Ronald R. Nepomuceno, Allison M. Rooks, Jeremy P. Hunt, Jill M. Ricono, Barbara Belli, and Robert C. Armstrong Abstract Fms-like tyrosine kinase 3 (FLT3) is implicated in the pathogenesis of acute myeloid leukemia (AML). FLT3-activating internal tandem duplication (ITD) mutations are found in approximately 30% of patients with AML and are associated with poor outcome in this patient population. Quizartinib (AC220) has previously been shown to be a potent and selective FLT3 inhibitor. In the current study, we expand on previous observations by showing that quizartinib potently inhibits the phosphorylation of FLT3 and downstream signaling molecules independent of FLT3 genotype, yet induces loss of viability only in cells expressing constitutively activated FLT3. We further show that transient exposure to quizartinib, whether in vitro or in vivo, leads to prolonged inhibition of FLT3 signaling, induction of apoptosis, and drastic reductions in tumor volume and pharmacodynamic endpoints. In vitro experiments suggest that these prolonged effects are mediated by slow binding kinetics that provide for durable inhibition of the kinase following drug removal/clearance. Together these data suggest quizartinib, with its unique combination of selectivity and potent/sustained inhibition of FLT3, may provide a safe and effective treatment against FLT3-driven leukemia. Mol Cancer Ther; 12(4); 1–10. Ó2013 AACR. Introduction downstream signaling cascades including the Ras/ Fms-like tyrosine kinase 3 (FLT3) is a member of the MAPK, PI3K/Akt, and STAT5 pathways (1, 9–13). -

Recombinant Human Thrombopoietin Promotes Hematopoietic
www.nature.com/scientificreports OPEN Recombinant human thrombopoietin promotes hematopoietic reconstruction after Received: 08 February 2015 Accepted: 06 July 2015 severe whole body irradiation Published: 25 September 2015 Chao Wang1,3,*, Bowen Zhang1,2,*, Sihan Wang1,2, Jing Zhang1,2, Yiming Liu1,2, Jingxue Wang1,2, Zeng Fan1,2, Yang Lv1,2, Xiuyuan Zhang1,2, Lijuan He1,2, Lin Chen1,2, Huanzhang Xia3, Yanhua Li1,2 & Xuetao Pei1,2 Recombinant human thrombopoietin (rHuTPO) is a drug that is used clinically to promote megakaryocyte and platelet generation. Here, we report the mitigative effect of rHuTPO (administered after exposure) against severe whole body irradiation in mice. Injection of rHuTPO for 14 consecutive days following exposure significantly improved the survival rate of lethally irradiated mice. RHuTPO treatment notably increased bone marrow cell density and LSK cell numbers in the mice after sub-lethal irradiation primarily by promoting residual HSC proliferation. In lethally irradiated mice with hematopoietic cell transplantation, rHuTPO treatment increased the survival rate and enhanced hematopoietic cell engraftment compared with the placebo treatment. Our observations indicate that recombinant human TPO might have a therapeutic role in promoting hematopoietic reconstitution and HSC engraftment. Accidental ionizing radiation exposure induces vital organ dysfunction syndromes in healthy individuals in radiological scenarios. The hematopoietic system is a radiosensitive organ that is highly suscepti- ble to damage1–3, and such damage can result in death. Clinically, radiation therapy or chemotherapy for patients with malignant diseases often leads to serious hematopoietic system damage. Modulation of the hematopoietic stem cell (HSC) population is considered to be key for realizing long-term sur- vival of patients. -
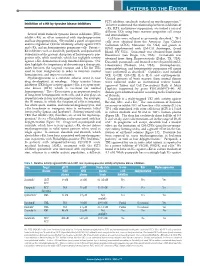
Letters to the Editor
LETTERS TO THE EDITOR FLT3 inhibitor, sorafenib, induced no myelosuppression.5,6 Inhibition of c-Kit by tyrosine kinase inhibitors To better understand the relationship between inhibition of c-Kit, FLT3, and marrow suppression, we studied a series of different TKIs using bone marrow progenitor cell assays Several small molecule tyrosine kinase inhibitors (TKIs) and immunoblots. inhibit c-Kit, an effect associated with myelosuppression Cell lines were cultured as previously described.2 TF-1 and hair depigmentation. We studied a panel of approved cells were obtained from the American Type Culture and investigational TKIs for inhibitory activity against FLT3 Collection (ATCC; Manassas, VA, USA) and grown in and c-Kit, and on hematopoietic progenitor cells. Potent c- RPMI supplemented with GM-CSF (Invitrogen, Grand Kit inhibitors such as dasatinib, pazopanib, and quizartinib Island, NY, USA). Quizartinib was obtained from Ambit demonstrated the greatest disruption of hematopoietic pro- Biosciences (San Diego, CA, USA). Crenolanib was genitor cells, while sorafenib, which has negligible activity obtained from Arog Pharmaceuticals (Dallas, TX, USA). against c-Kit, demonstrated only minimal disruption. Our Dasatinib, pazopanib, and imatinib were obtained from LC data highlight the importance of determining a therapeutic Laboratories (Woburn, MA, USA). Electrophoresis, index between the targeted receptor and c-Kit for TKIs immunoblotting, and hematopoietic progenitor cell assays used to treat malignancies in order to maintain normal were performed as described.2 Cytokines used included hematopoiesis and improve outcomes. SCF, G-CSF, GM-CSF, IL-3, IL-6, and erythropoietin. Myelosuppression is a common adverse event in new Unused portions of bone marrow from normal donors drug development in oncology.