Chapter 6: the Fermi Liquid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Density of States Information from Low Temperature Specific Heat
JOURNAL OF RESE ARC H of th e National Bureau of Standards - A. Physics and Chemistry Val. 74A, No.3, May-June 1970 Density of States I nformation from Low Temperature Specific Heat Measurements* Paul A. Beck and Helmut Claus University of Illinois, Urbana (October 10, 1969) The c a lcul ati on of one -electron d ensit y of s tate va lues from the coeffi cient y of the te rm of the low te mperature specifi c heat lin ear in te mperature is compli cated by many- body effects. In parti c ul ar, the electron-p honon inte raction may enhance the measured y as muc h as tw ofo ld. The e nha nce me nt fa ctor can be eva luat ed in the case of supe rconducting metals and a ll oys. In the presence of magneti c mo ments, add it ional complicati ons arise. A magneti c contribution to the measured y was ide ntifi e d in the case of dilute all oys and a lso of concentrated a lJ oys wh e re parasiti c antife rromagnetis m is s upe rim posed on a n over-a ll fe rromagneti c orde r. No me thod has as ye t bee n de vised to e valu ate this magne ti c part of y. T he separati on of the te mpera ture- li near term of the s pec ifi c heat may itself be co mpli cated by the a ppearance of a s pecific heat a no ma ly due to magneti c cluste rs in s upe rpa ramagneti c or we ak ly ferromagneti c a ll oys. -

Competition Between Paramagnetism and Diamagnetism in Charged
Competition between paramagnetism and diamagnetism in charged Fermi gases Xiaoling Jian, Jihong Qin, and Qiang Gu∗ Department of Physics, University of Science and Technology Beijing, Beijing 100083, China (Dated: November 10, 2018) The charged Fermi gas with a small Lande-factor g is expected to be diamagnetic, while that with a larger g could be paramagnetic. We calculate the critical value of the g-factor which separates the dia- and para-magnetic regions. In the weak-field limit, gc has the same value both at high and low temperatures, gc = 1/√12. Nevertheless, gc increases with the temperature reducing in finite magnetic fields. We also compare the gc value of Fermi gases with those of Boltzmann and Bose gases, supposing the particle has three Zeeman levels σ = 1, 0, and find that gc of Bose and Fermi gases is larger and smaller than that of Boltzmann gases,± respectively. PACS numbers: 05.30.Fk, 51.60.+a, 75.10.Lp, 75.20.-g I. INTRODUCTION mK and 10 µK, respectively. The ions can be regarded as charged Bose or Fermi gases. Once the quantum gas Magnetism of electron gases has been considerably has both the spin and charge degrees of freedom, there studied in condensed matter physics. In magnetic field, arises the competition between paramagnetism and dia- the magnetization of a free electron gas consists of two magnetism, as in electrons. Different from electrons, the independent parts. The spin magnetic moment of elec- g-factor for different magnetic ions is diverse, ranging trons results in the paramagnetic part (the Pauli para- from 0 to 2. -

Lecture 3: Fermi-Liquid Theory 1 General Considerations Concerning Condensed Matter
Phys 769 Selected Topics in Condensed Matter Physics Summer 2010 Lecture 3: Fermi-liquid theory Lecturer: Anthony J. Leggett TA: Bill Coish 1 General considerations concerning condensed matter (NB: Ultracold atomic gasses need separate discussion) Assume for simplicity a single atomic species. Then we have a collection of N (typically 1023) nuclei (denoted α,β,...) and (usually) ZN electrons (denoted i,j,...) interacting ∼ via a Hamiltonian Hˆ . To a first approximation, Hˆ is the nonrelativistic limit of the full Dirac Hamiltonian, namely1 ~2 ~2 1 e2 1 Hˆ = 2 2 + NR −2m ∇i − 2M ∇α 2 4πǫ r r α 0 i j Xi X Xij | − | 1 (Ze)2 1 1 Ze2 1 + . (1) 2 4πǫ0 Rα Rβ − 2 4πǫ0 ri Rα Xαβ | − | Xiα | − | For an isolated atom, the relevant energy scale is the Rydberg (R) – Z2R. In addition, there are some relativistic effects which may need to be considered. Most important is the spin-orbit interaction: µ Hˆ = B σ (v V (r )) (2) SO − c2 i · i × ∇ i Xi (µB is the Bohr magneton, vi is the velocity, and V (ri) is the electrostatic potential at 2 3 2 ri as obtained from HˆNR). In an isolated atom this term is o(α R) for H and o(Z α R) for a heavy atom (inner-shell electrons) (produces fine structure). The (electron-electron) magnetic dipole interaction is of the same order as HˆSO. The (electron-nucleus) hyperfine interaction is down relative to Hˆ by a factor µ /µ 10−3, and the nuclear dipole-dipole SO n B ∼ interaction by a factor (µ /µ )2 10−6. -

Fermi Surface of Copper Electrons That Do Not Interact
StringString theorytheory andand thethe mysteriousmysterious quantumquantum mattermatter ofof condensedcondensed mattermatter physics.physics. Jan Zaanen 1 String theory: what is it really good for? - Hadron (nuclear) physics: quark-gluon plasma in RIHC. - Quantum matter: quantum criticality in heavy fermion systems, high Tc superconductors, … Started in 2001, got on steam in 2007. Son Hartnoll Herzog Kovtun McGreevy Liu Schalm 2 Quantum critical matter Quark gluon plasma Iron High Tc Heavy fermions superconductors superconductors (?) Quantum critical Quantum critical 3 High-Tc Has Changed Landscape of Condensed Matter Physics High-resolution ARPES Magneto-optics Transport-Nernst effect Spin-polarized Neutron STM High Tc Superconductivity Inelastic X-Ray Scattering Angle-resolved MR/Heat Capacity ? Photoemission spectrum Hairy Black holes … 6 Holography and quantum matter But first: crash course in holography “Planckian dissipation”: quantum critical matter at high temperature, perfect fluids and the linear resistivity (Son, Policastro, …, Sachdev). Reissner Nordstrom black hole: “critical Fermi-liquids”, like high Tc’s normal state (Hong Liu, John McGreevy). Dirac hair/electron star: Fermi-liquids emerging from a non Fermi liquid (critical) ultraviolet, like overdoped high Tc (Schalm, Cubrovic, Hartnoll). Scalar hair: holographic superconductivity, a new mechanism for superconductivity at a high temperature (Hartnoll, Herzog,Horowitz) . 7 General relativity “=“ quantum field theory Gravity Quantum fields Maldacena 1997 = 8 Anti de Sitter-conformal -

Phys 446: Solid State Physics / Optical Properties Lattice Vibrations
Solid State Physics Lecture 5 Last week: Phys 446: (Ch. 3) • Phonons Solid State Physics / Optical Properties • Today: Einstein and Debye models for thermal capacity Lattice vibrations: Thermal conductivity Thermal, acoustic, and optical properties HW2 discussion Fall 2007 Lecture 5 Andrei Sirenko, NJIT 1 2 Material to be included in the test •Factors affecting the diffraction amplitude: Oct. 12th 2007 Atomic scattering factor (form factor): f = n(r)ei∆k⋅rl d 3r reflects distribution of electronic cloud. a ∫ r • Crystalline structures. 0 sin()∆k ⋅r In case of spherical distribution f = 4πr 2n(r) dr 7 crystal systems and 14 Bravais lattices a ∫ n 0 ∆k ⋅r • Crystallographic directions dhkl = 2 2 2 1 2 ⎛ h k l ⎞ 2πi(hu j +kv j +lw j ) and Miller indices ⎜ + + ⎟ •Structure factor F = f e ⎜ a2 b2 c2 ⎟ ∑ aj ⎝ ⎠ j • Definition of reciprocal lattice vectors: •Elastic stiffness and compliance. Strain and stress: definitions and relation between them in a linear regime (Hooke's law): σ ij = ∑Cijklε kl ε ij = ∑ Sijklσ kl • What is Brillouin zone kl kl 2 2 C •Elastic wave equation: ∂ u C ∂ u eff • Bragg formula: 2d·sinθ = mλ ; ∆k = G = eff x sound velocity v = ∂t 2 ρ ∂x2 ρ 3 4 • Lattice vibrations: acoustic and optical branches Summary of the Last Lecture In three-dimensional lattice with s atoms per unit cell there are Elastic properties – crystal is considered as continuous anisotropic 3s phonon branches: 3 acoustic, 3s - 3 optical medium • Phonon - the quantum of lattice vibration. Elastic stiffness and compliance tensors relate the strain and the Energy ħω; momentum ħq stress in a linear region (small displacements, harmonic potential) • Concept of the phonon density of states Hooke's law: σ ij = ∑Cijklε kl ε ij = ∑ Sijklσ kl • Einstein and Debye models for lattice heat capacity. -

Solid State Physics 2 Lecture 5: Electron Liquid
Physics 7450: Solid State Physics 2 Lecture 5: Electron liquid Leo Radzihovsky (Dated: 10 March, 2015) Abstract In these lectures, we will study itinerate electron liquid, namely metals. We will begin by re- viewing properties of noninteracting electron gas, developing its Greens functions, analyzing its thermodynamics, Pauli paramagnetism and Landau diamagnetism. We will recall how its thermo- dynamics is qualitatively distinct from that of a Boltzmann and Bose gases. As emphasized by Sommerfeld (1928), these qualitative di↵erence are due to the Pauli principle of electons’ fermionic statistics. We will then include e↵ects of Coulomb interaction, treating it in Hartree and Hartree- Fock approximation, computing the ground state energy and screening. We will then study itinerate Stoner ferromagnetism as well as various response functions, such as compressibility and conduc- tivity, and screening (Thomas-Fermi, Debye). We will then discuss Landau Fermi-liquid theory, which will allow us understand why despite strong electron-electron interactions, nevertheless much of the phenomenology of a Fermi gas extends to a Fermi liquid. We will conclude with discussion of electrons on the lattice, treated within the Hubbard and t-J models and will study transition to a Mott insulator and magnetism 1 I. INTRODUCTION A. Outline electron gas ground state and excitations • thermodynamics • Pauli paramagnetism • Landau diamagnetism • Hartree-Fock theory of interactions: ground state energy • Stoner ferromagnetic instability • response functions • Landau Fermi-liquid theory • electrons on the lattice: Hubbard and t-J models • Mott insulators and magnetism • B. Background In these lectures, we will study itinerate electron liquid, namely metals. In principle a fully quantum mechanical, strongly Coulomb-interacting description is required. -

Chapter 3 Bose-Einstein Condensation of an Ideal
Chapter 3 Bose-Einstein Condensation of An Ideal Gas An ideal gas consisting of non-interacting Bose particles is a ¯ctitious system since every realistic Bose gas shows some level of particle-particle interaction. Nevertheless, such a mathematical model provides the simplest example for the realization of Bose-Einstein condensation. This simple model, ¯rst studied by A. Einstein [1], correctly describes important basic properties of actual non-ideal (interacting) Bose gas. In particular, such basic concepts as BEC critical temperature Tc (or critical particle density nc), condensate fraction N0=N and the dimensionality issue will be obtained. 3.1 The ideal Bose gas in the canonical and grand canonical ensemble Suppose an ideal gas of non-interacting particles with ¯xed particle number N is trapped in a box with a volume V and at equilibrium temperature T . We assume a particle system somehow establishes an equilibrium temperature in spite of the absence of interaction. Such a system can be characterized by the thermodynamic partition function of canonical ensemble X Z = e¡¯ER ; (3.1) R where R stands for a macroscopic state of the gas and is uniquely speci¯ed by the occupa- tion number ni of each single particle state i: fn0; n1; ¢ ¢ ¢ ¢ ¢ ¢g. ¯ = 1=kBT is a temperature parameter. Then, the total energy of a macroscopic state R is given by only the kinetic energy: X ER = "ini; (3.2) i where "i is the eigen-energy of the single particle state i and the occupation number ni satis¯es the normalization condition X N = ni: (3.3) i 1 The probability -

Unconventional Hund Metal in a Weak Itinerant Ferromagnet
ARTICLE https://doi.org/10.1038/s41467-020-16868-4 OPEN Unconventional Hund metal in a weak itinerant ferromagnet Xiang Chen1, Igor Krivenko 2, Matthew B. Stone 3, Alexander I. Kolesnikov 3, Thomas Wolf4, ✉ ✉ Dmitry Reznik 5, Kevin S. Bedell6, Frank Lechermann7 & Stephen D. Wilson 1 The physics of weak itinerant ferromagnets is challenging due to their small magnetic moments and the ambiguous role of local interactions governing their electronic properties, 1234567890():,; many of which violate Fermi-liquid theory. While magnetic fluctuations play an important role in the materials’ unusual electronic states, the nature of these fluctuations and the paradigms through which they arise remain debated. Here we use inelastic neutron scattering to study magnetic fluctuations in the canonical weak itinerant ferromagnet MnSi. Data reveal that short-wavelength magnons continue to propagate until a mode crossing predicted for strongly interacting quasiparticles is reached, and the local susceptibility peaks at a coher- ence energy predicted for a correlated Hund metal by first-principles many-body theory. Scattering between electrons and orbital and spin fluctuations in MnSi can be understood at the local level to generate its non-Fermi liquid character. These results provide crucial insight into the role of interorbital Hund’s exchange within the broader class of enigmatic multiband itinerant, weak ferromagnets. 1 Materials Department, University of California, Santa Barbara, CA 93106, USA. 2 Department of Physics, University of Michigan, Ann Arbor, MI 48109, USA. 3 Neutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. 4 Institute for Solid State Physics, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany. -

High Pressure Band Structure, Density of States, Structural Phase Transition and Metallization in Cds
Chemical and Materials Engineering 5(1): 8-13, 2017 http://www.hrpub.org DOI: 10.13189/cme.2017.050102 High Pressure Band Structure, Density of States, Structural Phase Transition and Metallization in CdS J. Jesse Pius1, A. Lekshmi2, C. Nirmala Louis2,* 1Rohini College of Engineering, Nagercoil, Kanyakumari District, India 2Research Center in Physics, Holy Cross College, India Copyright©2017 by authors, all rights reserved. Authors agree that this article remains permanently open access under the terms of the Creative Commons Attribution License 4.0 International License Abstract The electronic band structure, density of high pressure studies due to the development of different states, metallization and structural phase transition of cubic designs of diamond anvil cell (DAC). With a modern DAC, zinc blende type cadmium sulphide (CdS) is investigated it is possible to reach pressures of 2 Mbar (200 GPa) using the full potential linear muffin-tin orbital (FP-LMTO) routinely and pressures of 5 Mbar (500 GPa) or higher is method. The ground state properties and band gap values achievable [3]. At such pressures, materials are reduced to are compared with the experimental results. The fractions of their original volumes. With this reduction in equilibrium lattice constant, bulk modulus and its pressure inter atomic distances; significant changes in bonding and derivative and the phase transition pressure at which the structure as well as other properties take place. The increase compounds undergo structural phase transition from ZnS to of pressure means the significant decrease in volume, which NaCl are predicted from the total energy calculations. The results in the change of electronic states and crystal structure. -
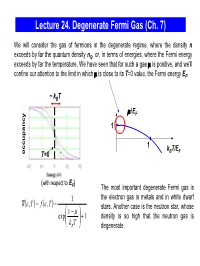
Lecture 24. Degenerate Fermi Gas (Ch
Lecture 24. Degenerate Fermi Gas (Ch. 7) We will consider the gas of fermions in the degenerate regime, where the density n exceeds by far the quantum density nQ, or, in terms of energies, where the Fermi energy exceeds by far the temperature. We have seen that for such a gas μ is positive, and we’ll confine our attention to the limit in which μ is close to its T=0 value, the Fermi energy EF. ~ kBT μ/EF 1 1 kBT/EF occupancy T=0 (with respect to E ) F The most important degenerate Fermi gas is 1 the electron gas in metals and in white dwarf nε()(),, T= f ε T = stars. Another case is the neutron star, whose ε⎛ − μ⎞ exp⎜ ⎟ +1 density is so high that the neutron gas is ⎝kB T⎠ degenerate. Degenerate Fermi Gas in Metals empty states ε We consider the mobile electrons in the conduction EF conduction band which can participate in the charge transport. The band energy is measured from the bottom of the conduction 0 band. When the metal atoms are brought together, valence their outer electrons break away and can move freely band through the solid. In good metals with the concentration ~ 1 electron/ion, the density of electrons in the electron states electron states conduction band n ~ 1 electron per (0.2 nm)3 ~ 1029 in an isolated in metal electrons/m3 . atom The electrons are prevented from escaping from the metal by the net Coulomb attraction to the positive ions; the energy required for an electron to escape (the work function) is typically a few eV. -

Chapter 13 Ideal Fermi
Chapter 13 Ideal Fermi gas The properties of an ideal Fermi gas are strongly determined by the Pauli principle. We shall consider the limit: k T µ,βµ 1, B � � which defines the degenerate Fermi gas. In this limit, the quantum mechanical nature of the system becomes especially important, and the system has little to do with the classical ideal gas. Since this chapter is devoted to fermions, we shall omit in the following the subscript ( ) that we used for the fermionic statistical quantities in the previous chapter. − 13.1 Equation of state Consider a gas ofN non-interacting fermions, e.g., electrons, whose one-particle wave- functionsϕ r(�r) are plane-waves. In this case, a complete set of quantum numbersr is given, for instance, by the three cartesian components of the wave vector �k and thez spin projectionm s of an electron: r (k , k , k , m ). ≡ x y z s Spin-independent Hamiltonians. We will consider only spin independent Hamiltonian operator of the type ˆ 3 H= �k ck† ck + d r V(r)c r†cr , �k � where thefirst and the second terms are respectively the kinetic and th potential energy. The summation over the statesr (whenever it has to be performed) can then be reduced to the summation over states with different wavevectork(p=¯hk): ... (2s + 1) ..., ⇒ r � �k where the summation over the spin quantum numberm s = s, s+1, . , s has been taken into account by the prefactor (2s + 1). − − 159 160 CHAPTER 13. IDEAL FERMI GAS Wavefunctions in a box. We as- sume that the electrons are in a vol- ume defined by a cube with sidesL x, Ly,L z and volumeV=L xLyLz. -

Extent of Fermi-Surface Reconstruction in the High-Temperature Superconductor Hgba2cuo4+Δ
Extent of Fermi-surface reconstruction in the high-temperature superconductor HgBa2CuO4+δ Mun K. Chana,1, Ross D. McDonalda, Brad J. Ramshawd, Jon B. Bettsa, Arkady Shekhterb, Eric D. Bauerc, and Neil Harrisona aPulsed Field Facility, National High Magnetic Field Laboratory, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA; bNational High Magnetic Field Laboratory, Florida State University, Tallahassee, Forida 32310, USA; cLos Alamos National Laboratory, Los Alamos, New Mexico, 87545, USA; dLaboratory of Atomic and Solid State Physics, Cornell University, Ithaca, NY 148.3, USA This manuscript was compiled on June 17, 2020 High magnetic fields have revealed a surprisingly small Fermi- change at TH ≈ 20 K. These results attest to the high-quality surface in underdoped cuprates, possibly resulting from Fermi- of the present crystals and confirm prior results indicating surface reconstruction due to an order parameter that breaks trans- Fermi-surface reconstruction in Hg1201 at similar doping lev- lational symmetry of the crystal lattice. A crucial issue concerns els (6,9, 10). the doping extent of this state and its relationship to the principal We have also found a peak in the planar resistivity ρ(T ) pseudogap and superconducting phases. We employ pulsed mag- at high fields, Figs.1C&D. ρ(T ) upturns at a temperature netic field measurements on the cuprate HgBa2CuO4+δ to identify coincident with the steep downturn in Rxy(T ), as shown in signatures of Fermi surface reconstruction from a sign change of the Fig.1D and SI Appendix Fig. S7. The common character- Hall effect and a peak in the temperature-dependent planar resistiv- istic temperatures suggest the features in ρ(T ) are related ity.